
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacology, Channabasweshwar Pharmacy College (Degree), Latur
Parkinson’s disease (PD) is a chronic and progressive neurodegenerative disorder characterized by selective degeneration of dopaminergic neurons in the substantia nigra pars compacta and depletion of striatal dopamine, resulting in a combination of motor and non-motor manifestations. Beyond the classical clinical features of resting tremor, bradykinesia, rigidity, and postural instability, PD is increasingly recognized as a multisystem disease involving cognitive dysfunction, psychiatric disturbances, autonomic impairment, sleep abnormalities, and gastrointestinal complications that substantially affect quality of life. The pathogenesis of PD is multifactorial and involves complex interactions among ?-synuclein aggregation, mitochondrial dysfunction, oxidative stress, neuroinflammation, and impairment of intracellular protein clearance pathways. Emerging evidence further supports the contribution of gut microbiota dysbiosis and gut–brain communication in disease initiation and progression. Genetic susceptibility, environmental exposures, and aging collectively influence neuronal vulnerability and disease heterogeneity. Despite major advances in understanding disease biology, currently available therapies remain largely symptomatic and fail to prevent neurodegeneration or alter long-term disease progression. Recent developments in biomarker discovery, molecular diagnostics, artificial intelligence, genomics, and precision medicine have opened new opportunities for earlier diagnosis and individualized therapeutic intervention. Novel strategies targeting ?-synuclein pathology, mitochondrial injury, neuroinflammation, and gut microbiota modulation represent promising directions for disease modification and improved clinical outcomes in PD management.
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by a combination of motor and non-motor manifestations resulting primarily from degeneration of dopaminergic neurons in the substantia nigra pars compacta and subsequent depletion of striatal dopamine [1]. Traditionally recognized by resting tremor, bradykinesia, rigidity, and postural instability, PD is now understood as a complex multisystem disorder involving cognitive, psychiatric, autonomic, and sensory dysfunctions that substantially impair quality of life [2]. PD is the second most prevalent neurodegenerative disorder after Alzheimer’s disease and represents a growing global public health challenge [3]. Disease prevalence increases markedly with age, particularly after 60 years, although hereditary and early-onset forms have also been identified [4]. Population aging, environmental exposures, and increasing life expectancy have contributed to a continuous rise in PD incidence worldwide over recent decades [5]. Consequently, considerable attention has been directed toward understanding disease mechanisms, identifying reliable biomarkers, and developing therapies capable of modifying disease progression rather than providing only symptomatic relief.
Neuropathologically, PD is characterized by progressive nigrostriatal degeneration together with intracellular deposition of misfolded α-synuclein aggregates in the form of Lewy bodies and Lewy neurites [6]. Under physiological conditions, α-synuclein participates in synaptic vesicle regulation; however, abnormal conformational changes promote aggregation, neuronal toxicity, and impairment of essential cellular pathways [7]. These aggregates disrupt mitochondrial activity, axonal transport, autophagy, and ubiquitin-proteasome function, ultimately leading to neuronal dysfunction and cell death [8]. Experimental and clinical evidence further suggests that pathological α-synuclein may spread through interconnected neural pathways in a prion-like manner, contributing to progressive involvement of both central and peripheral nervous systems [9].
Oxidative stress and mitochondrial dysfunction are also central contributors to PD pathogenesis. Dopaminergic neurons exhibit high metabolic demand and are particularly vulnerable to oxidative injury generated through dopamine metabolism and reactive oxygen species formation [10]. Excess oxidative stress induces lipid peroxidation, protein oxidation, and DNA damage, thereby accelerating neurodegeneration [11]. Mitochondrial abnormalities, especially impairment of complex I within the electron transport chain, further compromise cellular energy production and enhance oxidative damage [12]. Defective autophagy and proteasomal degradation additionally impair removal of damaged proteins and organelles, promoting intracellular accumulation of toxic aggregates [13]. Neuroinflammatory mechanisms have likewise gained significant attention in PD research. Activated microglia and astrocytes release pro-inflammatory cytokines and neurotoxic mediators that exacerbate neuronal injury [14]. Persistent inflammatory activation establishes a cycle in which oxidative stress, mitochondrial dysfunction, and inflammatory signaling reinforce one another, thereby contributing to progressive neuronal loss [15]. Current understanding therefore supports a multifactorial model in which protein aggregation, oxidative stress, mitochondrial impairment, and neuroinflammation interact closely during disease progression.
Although motor manifestations remain the clinical hallmark of PD, non-motor symptoms are increasingly recognized as major determinants of disability and reduced quality of life [16]. Many of these symptoms, including depression, anxiety, cognitive impairment, constipation, anosmia, sleep disturbances, fatigue, and autonomic dysfunction, may appear years before classical motor abnormalities become evident [17,18]. Recognition of these prodromal features has important implications for early diagnosis and therapeutic intervention. As disease advances, patients frequently develop gait impairment, motor fluctuations, dyskinesias, and cognitive decline that significantly increase long-term healthcare burden [19].
Recent studies have also emphasized the importance of the gut-brain axis in PD pathogenesis. According to the Braak hypothesis, abnormal α-synuclein aggregation may initially arise within the gastrointestinal tract or olfactory structures before propagating to the brain through vagal and interconnected neural pathways [20]. Alterations in gut microbiota composition have been associated with intestinal inflammation, immune dysregulation, increased gut permeability, and abnormal metabolite production, all of which may influence neurodegenerative processes [21]. These findings have stimulated growing interest in microbiome-based biomarkers and therapeutic strategies targeting early disease mechanisms.
Despite substantial advances in understanding PD biology, currently available treatments remain predominantly symptomatic [22]. Levodopa, dopamine agonists, monoamine oxidase inhibitors, and deep brain stimulation provide significant clinical benefit; however, long-term therapy is frequently complicated by dyskinesias, motor fluctuations, and declining therapeutic responsiveness [23]. Moreover, the absence of universally accepted early diagnostic biomarkers continues to limit intervention during preclinical stages of disease progression [24]. Consequently, considerable research efforts are now focused on disease-modifying approaches targeting α-synuclein aggregation, oxidative stress, mitochondrial dysfunction, neuroinflammation, and gut microbiota alterations.
Advances in molecular neuroscience, neuroimaging, genomics, and biomarker discovery have considerably expanded current understanding of PD pathophysiology [25]. Emerging therapeutic strategies including gene therapy, stem cell approaches, immunotherapy against α-synuclein, antioxidant agents, and neuroprotective interventions are under active investigation [26]. Although many of these approaches remain experimental, they offer promising directions for improving early diagnosis, slowing neurodegeneration, and enhancing long-term clinical management.
This review provides a comprehensive overview of Parkinson’s disease with emphasis on epidemiology, pathogenic mechanisms, neuropathological alterations, clinical manifestations, and emerging concepts involving oxidative stress, neuroinflammation, and the gut-brain axis. In addition, current therapeutic limitations, diagnostic challenges, and evolving disease-modifying strategies are critically discussed in the context of future translational and clinical applications.
2. Epidemiology of Parkinson’s Disease

Fig 1: Parkinson Disease Epidemiology
Parkinson’s disease (PD) is an increasingly important global neurological disorder whose prevalence continues to rise with population aging and increasing life expectancy. Epidemiological studies identify PD as one of the fastest-growing neurodegenerative diseases worldwide, contributing substantially to disability, healthcare expenditure, and socioeconomic burden. Disease occurrence varies according to age, sex, geography, environmental exposure, and genetic susceptibility [27].
2.1 Global Prevalence
PD is currently recognized as the second most common neurodegenerative disorder after Alzheimer’s disease. The global prevalence is estimated at approximately 0.3% in the general population and increases markedly with advancing age, affecting nearly 1-3% of individuals older than 60 years [27]. Current estimates indicate that more than 8 million individuals worldwide are living with PD, although the true burden is likely underestimated in low-resource regions because of underdiagnosis and limited neurological services [28].
Data from Global Burden of Disease (GBD) analyses demonstrate that the prevalence of PD has more than doubled over recent decades [36]. Population aging, increased survival, urbanization, and environmental exposures are considered major contributors to this rise. Improved clinical recognition and advances in diagnostic approaches have additionally increased case detection across many countries.
2.2 Incidence
The annual incidence of PD ranges from approximately 8 to 18 cases per 100,000 person-years, with considerable variation among populations and geographic regions [29]. Incidence increases progressively with age, particularly after the sixth decade of life, highlighting the strong association between aging and neurodegeneration.
Environmental and occupational factors also influence disease incidence. Chronic exposure to pesticides, herbicides, industrial solvents, and heavy metals has been associated with increased PD risk [31]. Several epidemiological studies have additionally linked rural residence and long-term well-water consumption with higher disease occurrence, although underlying mechanisms remain incompletely understood.
2.3 Age Distribution
Age is considered the most significant risk factor for PD. The average age at disease onset is approximately 60 years, and prevalence rises sharply among elderly populations [33]. Aging-related mitochondrial dysfunction, oxidative stress, impaired proteostasis, and reduced neuronal repair capacity are believed to increase vulnerability to neurodegeneration.
Early-onset Parkinson’s disease (EOPD), typically defined as onset before 50 years of age, accounts for approximately 5-10% of total cases [33]. Unlike late-onset sporadic PD, EOPD is more strongly associated with inherited mutations involving genes such as PARK2, PINK1, DJ-1, and LRRK2. These observations support the greater contribution of genetic susceptibility in younger patients.
2.4 Gender Differences
Sex-related differences are consistently observed in PD epidemiology, with males affected more frequently than females at an estimated ratio of approximately 1.5:1 [31]. Several biological and environmental mechanisms have been proposed to explain this disparity.
Estrogen may exert neuroprotective antioxidant and anti-inflammatory effects that reduce dopaminergic neuronal vulnerability in females. Differences in occupational exposure to environmental neurotoxins, immune responses, mitochondrial activity, and dopamine metabolism may also contribute to sex-based variability in disease susceptibility and progression.
2.5 Geographic Variation
Considerable geographic variation exists in PD prevalence and incidence. Higher rates are generally reported in Europe and North America, whereas lower prevalence has been observed in Africa and parts of Asia [32]. However, these differences may partly reflect disparities in healthcare access, diagnostic availability, life expectancy, epidemiological surveillance, and reporting accuracy rather than true biological variation alone.
Industrialized countries often demonstrate higher documented prevalence because of increased survival rates and better neurological healthcare systems. In contrast, underdiagnosis and limited access to specialists may contribute to lower reported prevalence in developing regions. Regional differences in environmental exposures, particularly pesticide use and industrial pollution, may further influence disease distribution.
2.6 Public Health Burden and Global Trends
The global burden of PD continues to increase rapidly and is projected to rise further in coming decades. Recent GBD analyses demonstrate substantial increases in PD-related disability-adjusted life years (DALYs), mortality, and healthcare costs worldwide [36]. Progressive motor impairment, cognitive decline, psychiatric manifestations, autonomic dysfunction, and loss of independence contribute significantly to long-term disability and caregiver burden.
The growing prevalence of PD is closely linked to demographic aging and global population transitions. Environmental pollution, sedentary lifestyles, metabolic disorders, and chronic exposure to neurotoxic agents may additionally contribute to increasing disease incidence. Consequently, PD is increasingly recognized as a major public health priority requiring improved epidemiological surveillance, earlier diagnosis, preventive strategies, and development of disease-modifying therapies.
3. Etiology and Risk Factors
Parkinson’s disease (PD) is a multifactorial neurodegenerative disorder resulting from complex interactions among genetic susceptibility, environmental exposures, aging-related cellular dysfunction, and lifestyle-associated factors. Although the precise etiology remains incompletely understood, substantial evidence indicates that α-synuclein aggregation, mitochondrial dysfunction, oxidative stress, neuroinflammation, and impaired protein degradation collectively contribute to disease pathogenesis [37,38]. These interconnected mechanisms contribute to considerable heterogeneity in clinical manifestations, disease progression, and therapeutic response.
3.1 Genetic Factors
Genetic factors contribute significantly to both familial and sporadic forms of PD. Approximately 5-10% of cases exhibit clear familial inheritance, whereas many sporadic cases are influenced by genetic polymorphisms that alter neuronal vulnerability [39]. Several genes associated with PD have been identified, including SNCA, LRRK2, PARKIN, PINK1, and DJ-1, each affecting pathways involved in neuronal survival and protein homeostasis. Mutations in the SNCA gene, which encodes α-synuclein, were among the earliest genetic abnormalities linked to familial PD. Under normal physiological conditions, α-synuclein regulates synaptic vesicle trafficking and neurotransmitter release; however, pathogenic mutations or gene multiplications promote protein misfolding and intracellular aggregation [40]. These aggregates accumulate as Lewy bodies and may spread through interconnected neural pathways in a prion-like manner, contributing to progressive neurodegeneration.
Mutations in leucine-rich repeat kinase 2 (LRRK2) are among the most common causes of autosomal dominant PD and are associated with abnormal kinase activity, impaired vesicular trafficking, mitochondrial dysfunction, and inflammatory signaling [39]. Similarly, mutations involving PARKIN, PINK1, and DJ-1 impair mitochondrial quality control, antioxidant defense, and ubiquitin-proteasome function, resulting in increased oxidative stress and neuronal vulnerability [38].
Although several PD-associated genes have been identified, genetic abnormalities alone rarely account for disease development. Current evidence supports a gene-environment interaction model in which inherited susceptibility enhances vulnerability to environmental toxins and aging-related cellular stress [41].
3.2 Environmental Factors
Environmental exposures are strongly implicated in sporadic PD. Epidemiological studies consistently associate chronic exposure to pesticides, herbicides, heavy metals, and industrial chemicals with increased disease risk [42,43].
Pesticides such as rotenone and paraquat have received considerable attention because they reproduce PD-like pathology in experimental models. These compounds impair mitochondrial complex I activity and promote excessive reactive oxygen species formation, leading to oxidative neuronal injury [44,46]. Dopaminergic neurons are particularly susceptible because of their high metabolic demand and intrinsic sensitivity to oxidative stress. Agricultural occupations, rural residence, and prolonged well-water consumption have also been associated with elevated PD risk, potentially reflecting chronic exposure to environmental neurotoxins [42]. Heavy metals including manganese and iron may additionally contribute to neurodegeneration through oxidative injury, mitochondrial dysfunction, and promotion of α-synuclein aggregation [43].
Industrial solvents and environmental pollutants have likewise been implicated in PD pathogenesis through mechanisms involving mitochondrial impairment, neuroinflammation, and disruption of cellular protein degradation pathways [41]. Head trauma has also emerged as a potential risk factor, as repetitive brain injury may initiate chronic inflammation and abnormal protein accumulation that contribute to dopaminergic neuronal degeneration [38].
Lifestyle-related factors may further influence disease susceptibility. Sedentary behavior and reduced physical activity may exacerbate oxidative stress and systemic inflammation, whereas regular exercise appears to reduce PD risk and improve neuronal resilience [50].
3.3 Aging and Cellular Vulnerability
Advancing age remains the strongest non-modifiable risk factor for PD, with most cases occurring after 60 years of age [43]. Aging contributes to cumulative cellular damage, impaired repair capacity, mitochondrial decline, and reduced neuronal resilience.
Dopaminergic neurons within the substantia nigra are particularly vulnerable because of their high metabolic activity, autonomous pacemaking properties, and increased production of reactive oxygen species [45]. Excessive oxidative stress damages proteins, lipids, nucleic acids, and mitochondrial DNA, thereby accelerating neurodegeneration [44]. Mitochondrial dysfunction is a central feature linking aging to PD pathogenesis. Aging mitochondria demonstrate impaired respiratory chain activity, reduced ATP synthesis, and increased oxidative burden [46]. Simultaneously, age-related decline in autophagy and ubiquitin-proteasome function promotes accumulation of misfolded proteins such as α-synuclein [47].
Neuroinflammation further contributes to neuronal injury. Activated microglia release pro-inflammatory cytokines and reactive intermediates that amplify oxidative stress and neuronal degeneration [48]. Persistent inflammatory activation may also facilitate additional α-synuclein aggregation and disease progression. Sex-related differences are also recognized in PD epidemiology, with males affected more frequently than females [49]. Estrogen is believed to exert antioxidant and anti-inflammatory effects that may partially protect dopaminergic neurons in women.
3.4 Protective Factors
Several factors have been associated with reduced PD risk, although underlying mechanisms remain incompletely understood. Regular physical activity is among the most consistently reported protective factors. Exercise improves mitochondrial efficiency, enhances neurotrophic signaling, increases cerebral perfusion, and reduces oxidative stress and inflammation [50].
Caffeine consumption has also demonstrated an inverse association with PD risk in multiple epidemiological studies [51]. Caffeine primarily acts through antagonism of adenosine A2A receptors, thereby modulating dopaminergic neurotransmission and inflammatory signaling pathways. Interestingly, cigarette smoking has repeatedly shown an inverse relationship with PD incidence, although smoking cannot be recommended because of its substantial adverse health consequences [52]. Nicotine may influence dopaminergic pathways and monoamine oxidase activity, although the mechanisms underlying this association remain uncertain.
Dietary antioxidants including flavonoids and polyphenols have additionally been investigated for potential neuroprotective effects [53]. These compounds may reduce oxidative stress, suppress inflammation, and limit α-synuclein aggregation, although clinical evidence remains inconsistent.
Overall, Parkinson’s disease develops through dynamic interactions among genetic predisposition, environmental exposures, aging-related degeneration, and lifestyle-associated influences. Understanding these etiological mechanisms is essential for identifying high-risk populations and developing preventive as well as disease-modifying therapeutic strategies.
4. Pathophysiology of Parkinson’s Disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized primarily by degeneration of dopaminergic neurons within the substantia nigra pars compacta (SNpc) and intracellular accumulation of misfolded α-synuclein aggregates [54,55]. Current evidence indicates that PD is a multifactorial disorder involving interconnected mechanisms including mitochondrial dysfunction, oxidative stress, impaired protein degradation, neuroinflammation, excitotoxicity, and disturbances in gut-brain signaling [54-56]. The pathological hallmarks of PD include nigrostriatal degeneration and formation of Lewy bodies composed predominantly of aggregated α-synuclein [57,58]. In addition to motor abnormalities, widespread neurodegeneration contributes to cognitive impairment, autonomic dysfunction, psychiatric manifestations, and gastrointestinal disturbances [56,59].
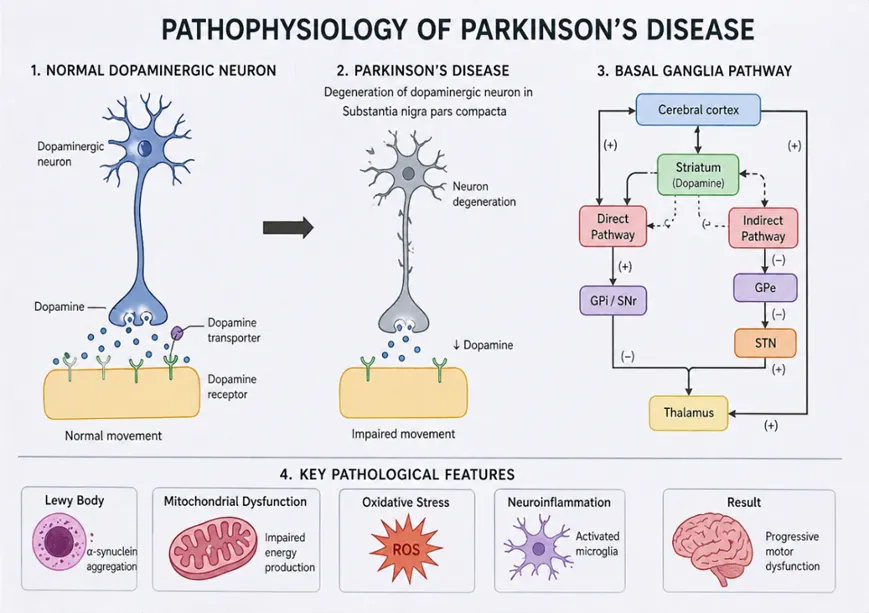
Fig 2: Pathophysiology of Parkinsons Disease
4.1 Dopaminergic Neurodegeneration
Degeneration of dopaminergic neurons within the SNpc represents the central neuropathological feature of PD [60]. These neurons regulate voluntary movement through modulation of basal ganglia circuits. Dopamine depletion disrupts the balance between direct and indirect motor pathways, resulting in excessive inhibitory output from the basal ganglia and reduced thalamocortical activation [61,62]. Clinically, this manifests as bradykinesia, rigidity, resting tremor, and postural instability.

Fig.3 Basal Ganglia Circuitry: Normal Function and Parkinson’s Disease Mechanisms
Several factors contribute to selective neuronal vulnerability. SNpc neurons exhibit extensive axonal arborization, high metabolic demand, autonomous pacemaking activity, and sustained calcium influx, making them particularly susceptible to oxidative and mitochondrial stress [63]. Dopamine metabolism additionally generates reactive oxygen species (ROS) and toxic dopamine quinones capable of inducing cellular injury [64]. Excess glutamatergic stimulation and calcium dysregulation further promote excitotoxicity and apoptotic neuronal death [65].
4.2 α-Synuclein Aggregation and Lewy Bodies
Aggregation of α-synuclein is a defining pathological event in PD [57]. Under physiological conditions, α-synuclein participates in synaptic vesicle trafficking and neurotransmitter release [66]. However, abnormal conformational changes promote formation of toxic oligomers and fibrillar aggregates [67].
Accumulation of aggregated α-synuclein within neurons leads to formation of Lewy bodies and Lewy neurites, characteristic pathological hallmarks of PD [57,68]. Misfolded α-synuclein disrupts synaptic transmission, mitochondrial activity, vesicular transport, and membrane integrity [69]. Soluble oligomeric forms are considered particularly neurotoxic because they impair neuronal homeostasis and cellular signaling pathways.
Experimental evidence further suggests that pathological α-synuclein propagates between interconnected neuronal networks in a prion-like manner [56,67]. Misfolded α-synuclein may act as a template that induces aggregation of native protein, thereby facilitating progressive spread of pathology across different brain regions.
4.3 Oxidative Stress and Mitochondrial Dysfunction
Oxidative stress and mitochondrial dysfunction are central contributors to PD pathogenesis [64,70]. Impairment of mitochondrial respiratory chain complex I reduces ATP synthesis and enhances ROS generation [70,71]. Environmental toxins such as MPTP and rotenone reproduce parkinsonian neurodegeneration experimentally through inhibition of complex I activity. Excessive ROS production damages proteins, lipids, nucleic acids, and mitochondrial DNA, thereby promoting neuronal dysfunction and apoptosis [64,72]. Dopaminergic neurons are especially vulnerable because dopamine metabolism itself produces reactive intermediates capable of inducing oxidative injury. Iron accumulation within the substantia nigra further amplifies oxidative stress through free radical generation [72].
Mitochondrial dysfunction also impairs calcium homeostasis and activates stress-related apoptotic pathways [70]. Defective mitophagy and impaired mitochondrial quality control permit accumulation of dysfunctional mitochondria, perpetuating oxidative damage and neuronal degeneration.
4.4 Neuroinflammation
Neuroinflammation is increasingly recognized as a major contributor to PD progression [73]. Activated microglia are abundant within the substantia nigra of PD patients and release pro-inflammatory cytokines, nitric oxide, and reactive oxygen intermediates [73,74]. Inflammatory mediators including TNF-α, IL-1β, and IL-6 exacerbate oxidative stress and neuronal injury.
Misfolded α-synuclein itself functions as an immunogenic stimulus capable of activating microglial inflammatory pathways [69,74]. Persistent microglial activation establishes a self-perpetuating cycle in which neurodegeneration promotes inflammation and inflammation accelerates further neuronal damage.
Astrocytes also contribute to inflammatory responses through release of cytokines and reactive nitrogen species [73]. Emerging evidence additionally suggests involvement of adaptive immune responses in PD pathogenesis.
4.5 Impaired Protein Clearance Systems
Efficient protein quality control is essential for neuronal survival. In PD, dysfunction of the ubiquitin-proteasome system and autophagy-lysosomal pathway contributes to accumulation of misfolded proteins and damaged organelles [75,76].
Autophagy plays a critical role in degradation of aggregated proteins and dysfunctional mitochondria. However, abnormal α-synuclein impairs lysosomal activity and disrupts autophagic flux, leading to progressive intracellular accumulation of toxic protein aggregates [75]. Failure of these degradative pathways further aggravates mitochondrial dysfunction, oxidative stress, and apoptotic neuronal death.
4.6 Gut-Brain Axis
Emerging evidence suggests that PD pathology may originate within the gastrointestinal tract before spreading to the central nervous system [77]. The gut-brain axis involves bidirectional communication among the enteric nervous system, intestinal microbiota, immune pathways, and the brain [78]. Gastrointestinal dysfunction frequently precedes motor manifestations by several years, indicating early involvement of enteric neurons [77]. α-Synuclein aggregates have been identified in intestinal tissues and vagal nerve fibers during prodromal stages of PD [58,77]. According to Braak’s hypothesis, pathological α-synuclein may spread retrogradely from the gastrointestinal tract to the brain through the vagus nerve.
Gut microbiota dysbiosis contributes to intestinal inflammation, increased gut permeability, and systemic immune activation [78,79]. Translocation of bacterial endotoxins such as lipopolysaccharide may activate microglia and promote neuroinflammation. Altered microbial metabolites may additionally influence neurotransmitter synthesis and α-synuclein aggregation. Epidemiological evidence demonstrating reduced PD risk following vagotomy further supports the concept of gut-to-brain pathological propagation [80].
5. Clinical Manifestations of Parkinson’s Disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by a wide range of motor and non-motor manifestations that collectively impair functional independence and quality of life. The disease primarily results from degeneration of dopaminergic neurons in the substantia nigra pars compacta with consequent disruption of basal ganglia circuitry. In addition, involvement of non-dopaminergic systems contributes to the heterogeneous clinical phenotype. Importantly, non-motor symptoms often precede motor manifestations, indicating early multisystem neurodegeneration beyond the nigrostriatal pathway [81,82].
5.1 Motor Symptoms
Motor features remain the core clinical presentation of PD and form the basis for diagnosis. The classical triad includes resting tremor, bradykinesia, and rigidity, with postural instability typically emerging in later stages [83].
Resting tremor is usually asymmetric and most evident at rest, commonly described as a “pill-rolling” movement. While not always functionally disabling in early stages, it can interfere with fine motor tasks and daily activities [83]. Bradykinesia is the most characteristic feature of PD and refers to slowness of movement with progressive reduction in amplitude during repetitive actions. It manifests clinically as hypomimia, micrographia, reduced arm swing, and gait impairment including shuffling and freezing, significantly contributing to disability [83].
Rigidity presents as increased resistance to passive movement and may be described as lead-pipe or cogwheel rigidity. It contributes to stiffness, pain, and reduced mobility, often progressing to postural deformities [83] Postural instability generally appears in advanced stages and is associated with impaired balance reflexes and increased risk of falls, fractures, and loss of independence [81].
5.2 Non-Motor Symptoms
Non-motor symptoms are now recognized as major determinants of disease burden and quality of life. These may precede motor onset by several years and reflect widespread neurodegeneration involving multiple neurotransmitter systems [82].
Cognitive impairment ranges from mild executive dysfunction to Parkinson’s disease dementia, affecting attention, memory, and visuospatial abilities, and contributing to loss of independence [84]. Psychiatric manifestations such as depression, anxiety, apathy, and hallucinations are common and may occur independently of motor severity. Visual hallucinations are particularly associated with advanced disease and dopaminergic therapy [85].
Sleep disturbances, including insomnia, excessive daytime sleepiness, and REM sleep behavior disorder (RBD), are frequent. RBD is especially important as a prodromal marker of synucleinopathy and may precede motor symptoms by years [84].
Autonomic dysfunction includes constipation, orthostatic hypotension, urinary disturbances, and sexual dysfunction. Gastrointestinal symptoms are often early features, supporting involvement of the gut-brain axis [82]. Sensory symptoms such as hyposmia are common early manifestations and may appear before motor symptoms. Chronic pain and sensory discomfort further contribute to reduced quality of life [84].
5.3 Disease Progression
PD follows a chronic progressive course with gradual worsening of both motor and non-motor features. Early disease is typically unilateral, while advanced stages involve bilateral motor impairment, postural instability, cognitive decline, and severe autonomic dysfunction [81].
Disease severity is commonly assessed using the Hoehn and Yahr staging system, ranging from unilateral involvement (Stage I) to complete dependence (Stage V) [83]. Progression is associated with increasing treatment complexity, reduced dopaminergic responsiveness, and higher risk of complications such as falls, aspiration, and dementia.
Ultimately, progressive neurodegeneration leads to significant disability, caregiver burden, and increased mortality. Early recognition and multidisciplinary management remain essential to improve long-term outcomes [81].
6. Diagnosis of Parkinson’s Disease
6.1 Clinical Diagnosis
Parkinson’s disease (PD) is primarily diagnosed on clinical grounds, with bradykinesia as the essential diagnostic requirement accompanied by either resting tremor or rigidity [86]. Early disease typically presents asymmetrically with progressive involvement of gait, reduced arm swing, hypomimia, micrographia, and later postural instability [87]. Importantly, non-motor symptoms such as constipation, hyposmia, REM sleep behavior disorder, depression, and autonomic dysfunction may precede motor onset, representing key prodromal markers [88].
Neurological examination remains essential for distinguishing idiopathic PD from atypical and secondary parkinsonian syndromes. Features such as early falls, vertical gaze palsy, cerebellar signs, pyramidal tract involvement, or rapid progression suggest alternative diagnoses [89]. Clinical accuracy is limited in early disease due to overlap with essential tremor, vascular parkinsonism, and drug-induced forms [86].
Levodopa responsiveness provides supportive diagnostic evidence, with significant improvement in bradykinesia and rigidity indicating nigrostriatal dysfunction [90]. However, responsiveness is not fully specific, as early atypical cases may transiently respond.
6.2 Diagnostic Criteria
The UK Brain Bank criteria improved diagnostic consistency but emphasized motor features and lacked sensitivity for prodromal disease [91]. The Movement Disorder Society (MDS) criteria introduced a structured framework incorporating supportive features, exclusion criteria, and non-motor biomarkers [92]. Diagnosis is classified as “clinically established” or “probable PD” based on weighted clinical features.
Definitive diagnosis remains neuropathological, requiring α-synuclein-positive Lewy pathology with nigral degeneration [93].
6.3 Neuroimaging
MRI is primarily used to exclude structural lesions and atypical parkinsonism rather than confirm PD [94]. Advanced MRI techniques may reveal subtle nigral changes but are not yet diagnostic. Dopamine transporter imaging (DaTscan) evaluates presynaptic dopaminergic integrity and helps differentiate degenerative parkinsonism from essential tremor [95]. However, it cannot reliably distinguish PD from atypical degenerative syndromes.
PET and SPECT imaging provide functional assessment of dopaminergic pathways and metabolic activity but are limited by cost and accessibility [96].
6.4 Biomarkers and Emerging Diagnostics
CSF biomarkers such as α-synuclein, tau, and neurofilament light chain show potential but lack standardization [97]. Reduced total α-synuclein with elevated oligomeric forms is frequently reported. α-synuclein seed amplification assays (SAA) have significantly improved diagnostic sensitivity and may enable preclinical detection [98]. Emerging evidence supports their utility in both CSF and peripheral tissues.
Machine learning approaches using gait, speech, imaging, and wearable sensor data demonstrate promising diagnostic accuracy but require further validation for clinical integration [99].
6.5 Differential Diagnosis
Key differentials include essential tremor, which lacks bradykinesia, and drug-induced parkinsonism, which is often reversible [100]. Atypical parkinsonian disorders such as multiple system atrophy, progressive supranuclear palsy, corticobasal degeneration, and dementia with Lewy bodies show additional neurological features and poorer levodopa response [89].
7. Therapeutic Management
7.1 Pharmacological Therapy
Levodopa remains the most effective symptomatic therapy for PD, improving bradykinesia and rigidity [101]. Chronic use, however, is associated with motor fluctuations and dyskinesias due to disease progression and pulsatile dopaminergic stimulation [101].
Dopamine agonists provide adjunctive benefit but are associated with neuropsychiatric adverse effects including hallucinations and impulse control disorders [102]. MAO-B inhibitors and COMT inhibitors prolong dopaminergic activity and reduce “off” periods [103]. Amantadine is useful for managing levodopa-induced dyskinesias [104].
7.2 Non-Pharmacological Therapy
Physiotherapy improves gait, balance, and fall risk, while occupational therapy supports daily functioning. Speech therapy addresses dysarthria and dysphagia. Regular exercise has additional neuroprotective and functional benefits [105].
7.3 Surgical Therapy
Deep brain stimulation (DBS) of the subthalamic nucleus or globus pallidus interna is effective in advanced PD with motor fluctuations [101]. It reduces medication requirements but does not halt neurodegeneration and may cause neuropsychiatric or speech-related complications [102].
8. Emerging Therapeutic Approaches
Disease-modifying strategies focus on α-synuclein aggregation, mitochondrial dysfunction, and neuroinflammation [97]. Gene therapy and stem cell-based approaches aim to restore dopaminergic function but remain experimental with unresolved long-term safety concerns [103]. α-synuclein immunotherapy has shown target engagement in trials but lacks consistent clinical benefit [98]. GLP-1 receptor agonists and mitochondrial stabilizers are promising neuroprotective candidates [104].
Nanotechnology-based delivery systems may enhance blood-brain barrier penetration and drug targeting, although translational barriers remain [99]. Gut microbiome modulation represents a novel therapeutic direction supported by increasing mechanistic evidence linking dysbiosis to PD pathogenesis [88]. Precision medicine and AI-based stratification approaches are expected to improve individualized therapy selection in future clinical practice [99].
9. FUTURE PERSPECTIVES
Future research in Parkinson’s disease (PD) is increasingly focused on shifting from symptomatic management toward early diagnosis, disease modification, and precision-guided intervention. A key priority is the development of robust biomarkers capable of detecting prodromal neurodegeneration prior to irreversible dopaminergic loss. Multi-source biomarker strategies incorporating cerebrospinal fluid, blood-based analytes, neuroimaging signatures, and peripheral tissue markers are being actively investigated to improve early diagnostic accuracy and disease stratification [106].
Among these, α-synuclein seeding amplification assays have shown particular promise in identifying misfolded protein aggregates at preclinical stages, supporting their potential role in early molecular diagnosis. In parallel, growing evidence implicates inflammatory mediators, mitochondrial dysfunction markers, and gut microbiome-derived signatures as complementary biomarker candidates that may better reflect disease heterogeneity and progression dynamics [106].
Advances in genomics and systems biology are accelerating the transition toward personalized medicine in PD. Stratification based on pathogenic variants such as LRRK2, SNCA, and GBA may enable individualized risk prediction and therapy selection, while also improving prognostic modeling [107]. Concurrently, artificial intelligence (AI) and machine learning approaches are being integrated with multimodal datasets, including clinical features, neuroimaging, speech patterns, and wearable sensor outputs, to enhance diagnostic precision and enable continuous digital phenotyping of disease progression [108].
Therapeutically, there is growing consensus that effective disease modification will likely require multi-target approaches addressing α-synuclein aggregation, neuroinflammation, lysosomal dysfunction, oxidative stress, and mitochondrial impairment simultaneously. Single-pathway interventions have thus far shown limited success, underscoring the need for combinatorial or network-based therapeutic strategies [109]. Additionally, digital health technologies and wearable monitoring systems are expected to transform PD management by enabling real-time symptom tracking, adaptive treatment adjustment, and decentralized clinical trial designs [108].
CONCLUSION
Parkinson’s disease is a complex neurodegenerative disorder driven by interacting molecular and cellular mechanisms, including dopaminergic neuronal loss, α-synuclein aggregation, mitochondrial dysfunction, oxidative stress, and impaired protein homeostasis. Clinically, it manifests through a broad spectrum of motor and non-motor symptoms that progressively impair functional independence and quality of life, imposing a significant socioeconomic burden. Current therapeutic strategies remain primarily symptomatic and do not alter disease trajectory. However, advances in molecular pathology, biomarker discovery, and computational neuroscience are reshaping the conceptual framework of PD toward earlier detection and mechanism-based intervention. Integration of precision medicine, AI-assisted diagnostics, and multi-target therapeutic development holds substantial promise for improving long-term outcomes.
Future progress will depend on bridging translational gaps between experimental findings and clinical application, with an emphasis on early intervention, individualized treatment strategies, and continuous disease monitoring. Collectively, these developments are expected to shift Parkinson’s disease management toward a predictive, preventive, and personalized medical model.
REFERENCES


Shubham Turewale, Dr. Padmaja Giram, Mahesh Manke, Rushikesh Choudhari, Kiran Surwase, Parkinson's Disease Beyond Dopamine: Integrating Molecular Pathogenesis, Biomarker Discovery, and Emerging Therapeutic Strategies, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 1088-1105, https://doi.org/10.5281/zenodo.21217551
 10.5281/zenodo.21217551
10.5281/zenodo.21217551