
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,3 Fergana Medical Institute Of Public Health, Uzbekistan
2 Dr. Rajendra Gode Nursing Institute of Buldhana, India
Protein–protein interactions (PPIs) constitute the fundamental molecular framework through which cells coordinate their biological activities, ranging from gene expression and metabolic regulation to signal transduction and structural organization. This comprehensive review synthesizes current knowledge on the molecular architecture of PPIs, their classification, detection methodologies, and their roles within cellular networks including signaling cascades, metabolic pathways, cytoskeletal assemblies, and ribosomal machinery. We examine the physicochemical forces that govern binding specificity and affinity—including hydrophobic contacts, electrostatic interactions, hydrogen bonding, and van der Waals forces—and explore how transient versus permanent interaction modes determine functional outcomes. The review further addresses the expanding interactome concept across model organisms, including plants, and discusses the role of computational and artificial intelligence (AI)-driven approaches—most notably AlphaFold2 and cryo-electron microscopy (cryo-EM)—in transforming structural proteomics. The therapeutic implications of PPI modulation are evaluated with emphasis on small molecules, peptides, and monoclonal antibodies targeting pathologically relevant complexes in cancer, neurodegeneration, and metabolic disorders. Taken together, the evidence underscores that elucidating and therapeutically manipulating PPIs represents one of the most promising frontiers in modern biomedical science.
Proteins are multifunctional macromolecules that serve as the principal catalysts, structural scaffolds, signaling messengers, and molecular machines of all living systems [1,2]. Protein–protein interactions (PPIs) are not merely accessory phenomena; they are the molecular language through which cellular events are orchestrated with extraordinary precision and spatiotemporal control [3,4].
The concept of the interactome—the complete set of PPIs occurring within a given organism or cell type—has transformed our understanding of biology from a reductionist paradigm to a systems-level perspective [5,6]. Rather than acting as isolated entities, proteins are embedded in elaborate networks of physical and functional associations, the topology and dynamics of which determine cellular phenotype, developmental fate, and pathological susceptibility [7,8].
Virtually every cellular function—including DNA replication, RNA splicing, protein translation, membrane transport, cytoskeletal dynamics, and apoptosis—depends on the precise assembly and disassembly of multiprotein complexes [9,10]. High-throughput experimental approaches such as yeast two-hybrid (Y2H) screening, affinity purification combined with mass spectrometry (AP-MS), and proximity labeling have collectively mapped thousands of interactions across diverse organisms [11,12]. Concurrently, deep learning structural predictors such as AlphaFold2 have exponentially expanded the catalogued interactome [13,14].
The therapeutic relevance of PPIs is increasingly appreciated. Small molecules, peptide mimetics, and biologic agents that selectively inhibit or stabilize pathological PPIs have entered clinical development for cancer, infectious disease, neurodegeneration, and immune disorders [15,16]. Landmark examples include venetoclax (ABT-199), a BCL-2 inhibitor approved for leukemia, and MDM2–p53 inhibitors under clinical evaluation in solid tumors [17,18].
The present review provides a comprehensive synthesis of PPI biology. We discuss the structural and physicochemical basis of protein recognition, classification and functional significance of PPI types, state-of-the-art detection methodologies, PPI network organization in signaling and metabolic contexts, and the emerging role of AI and cryo-EM in structural proteomics, concluding with a critical assessment of the challenges and opportunities in targeting PPIs for therapeutic benefit.
2. Definition, Classification, and Biological Context of Protein–Protein Interactions
2.1 Defining Protein–Protein Interactions
A protein–protein interaction is defined as a physical contact between two or more protein molecules resulting from specific non-covalent forces and producing a biologically meaningful outcome [19]. This definition excludes coincidental contacts arising from random molecular crowding, as well as indirect associations mediated solely by DNA, RNA, or small-molecule cofactors [20]. Within the ribosome or spliceosome, many subunits are spatially adjacent yet interact directly only with a subset of neighbors, requiring experimental evidence of direct contact via co-immunoprecipitation, surface plasmon resonance, or structural characterization [21,22].
Context dependency is a defining feature of PPIs. A given interaction may occur exclusively in specific cell types, during particular phases of the cell cycle, or in response to defined extracellular stimuli [23]. Post-translational modifications (PTMs)—including phosphorylation, ubiquitination, acetylation, methylation, and SUMOylation—dynamically regulate PPI engagement by creating, destroying, or remodeling binding surfaces [24,25].
2.2 Functional Classification of PPIs
PPIs are most usefully classified along several axes: (i) stability (permanent vs. transient), (ii) partner specificity (homomeric vs. heteromeric), (iii) obligateness (obligate vs. non-obligate), and (iv) biological role (structural, catalytic, regulatory, or scaffolding) [26,27]. Permanent complexes—including the F-type ATP synthase, cytochrome c oxidase, and the proteasome regulatory particle—are stable under physiological conditions [28,29]. Transient complexes assemble only under specific conditions and disassemble thereafter, allowing dynamic signal relay [30].
Obligate interactions occur between partners structurally and functionally dependent on one another, whereas non-obligate interactions involve independently folded partners that associate conditionally [31,32]. Homomeric complexes arise from identical chains interacting, while heteromeric complexes—the majority of signaling and structural assemblies—involve distinct polypeptide chains [33,34].
2.3 Co-interacting, Correlated, and Co-localized Proteins
A useful three-level taxonomy distinguishes direct co-interacting proteins (forming physical complexes), correlated proteins (participating in shared pathways without direct contact), and co-localized proteins (residing in the same compartment but not necessarily interacting) [35,36]. This hierarchy is essential for interpreting large-scale interactome datasets, which frequently conflate these levels. Co-localized proteins share membrane compartments or cytoskeletal scaffolds but may function in entirely distinct processes [37].
3. Molecular Basis of Protein–Protein Interactions and Binding Site Architecture
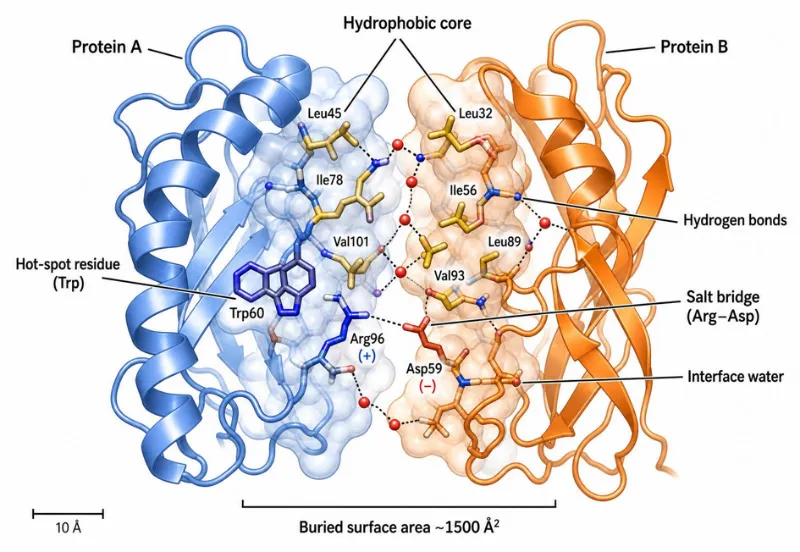
Figure 1. Molecular architecture of a protein–protein interaction interface. Two ribbon-diagram protein domains (Protein A, blue; Protein B, orange) dock through a buried surface area of \~1,500 Ų. The interface is stabilized by a hydrophobic core, hot-spot residues (Trp), salt bridges (Arg–Asp), hydrogen bonds, van der Waals contacts, and interface-bridging water molecules.
3.1 Structural Features of Protein Binding Interfaces
Protein–protein interfaces possess distinctive structural characteristics that differentiate them from the remainder of the protein surface [38]. Interfaces typically encompass 750–2,000 Ų of buried solvent-accessible surface area per chain and are enriched in aromatic residues (Trp, Tyr, Phe), hydrophobic residues (Leu, Ile, Val), and charged residues participating in salt bridges [39,40]. High-resolution structural analyses reveal heterogeneous interface topography featuring pockets, protrusions, and indentations; geometric complementarities are highest in permanent and obligate complexes and lowest in antibody–antigen contacts [41].
A critical concept is that of hot spots—interface residues contributing disproportionately to binding free energy despite comprising a minority of interface area [42,43]. Computational alanine scanning and experimental mutagenesis establish that hot spot residues cluster at the interface center, are conserved across homologs, and are surrounded by peripheral residues that maintain geometric complementarity while contributing little to affinity [44,45].
3.2 Physicochemical Forces Governing Binding
The thermodynamic driving force for PPI formation is provided by the net balance of enthalpy and entropy changes upon complex assembly [46]. Hydrophobic burial is the dominant enthalpic contributor: transfer of apolar surface from solvent to the buried interface is thermodynamically favorable owing to entropic gain from released water molecules [47]. Permanent and obligate complexes typically exhibit more extensive hydrophobic cores than transient assemblies [48].
Electrostatic interactions—ion pairs (salt bridges), charge–dipole contacts, and Coulombic forces—contribute substantially to binding specificity and orientation [49,50]. Hydrogen bonds, averaging one per 100–200 Ų of interface area, provide directional specificity and enthalpic stabilization [51]. Approximately 30% of protein–protein interfaces are bridged by ordered water molecules that hydrogen-bond to both partners, contributing to specificity while maintaining interfacial flexibility [52,53].
3.3 Conformational Dynamics and Induced-Fit Binding
The classical lock-and-key model of protein recognition has been substantially revised. Two primary mechanisms are characterized: induced fit, where the unbound protein rearranges upon partner encounter; and conformational selection, where a minor binding-competent conformation is selectively stabilized upon partner binding [54,55]. Intrinsically disordered proteins (IDPs) represent an extreme case where substantial folding occurs as a consequence of partner binding—termed folding-upon-binding—offering kinetic advantages including large accessible binding surfaces and the capacity to engage multiple partners [56,57].
Computational molecular dynamics simulations reveal that normal mode motions of the unbound protein often correlate with the observed conformational transitions upon binding, suggesting that the conformational change trajectory is encoded in the intrinsic flexibility of the unbound structure [58]. These insights are practically important for computational docking and virtual screening campaigns targeting PPIs.
4. Transient and Permanent Protein–Protein Interactions: Structural and Functional Perspectives
4.1 Criteria for Classification
The delineation of transient from permanent protein complexes carries profound biophysical and functional implications [59]. Permanent complexes—exemplified by the proteasome, ribosome, and respiratory chain supercomplexes—are characterized by extensive hydrophobic interfaces, high thermodynamic stability, and slow dissociation rates [60]. Transient complexes associate and dissociate on timescales from milliseconds to minutes, with dissociation constants (Kd) spanning picomolar to millimolar ranges [61].
A further subdivision distinguishes weak transient complexes—homodimeric enzymes in equilibrium between monomeric and dimeric states—from strong transient complexes that switch quaternary state only upon a defined trigger such as ligand binding, phosphorylation, or nucleotide exchange [62]. The heterotrimeric G-protein is a paradigmatic strong transient complex: the Gα–Gβγ heterotrimer dissociates specifically upon GTP binding to Gα following receptor activation [63].
4.2 Structural Determinants of Interface Stability
Comparative structural analyses reveal that permanent interfaces are larger, more hydrophobic, and more planar than transient interfaces [64]. Transient interfaces are more polar, smaller, and less geometrically complementary, consistent with requirements for rapid and specific yet reversible association [65]. Molecular weight asymmetry between partners is a particularly predictive feature distinguishing permanent from transient complexes, outperforming interface area, hydrophobicity indices, and hydrogen bond counts [66].
Thermodynamic characterization of transient complexes employs size exclusion chromatography, analytical ultracentrifugation, isothermal titration calorimetry, and fluorescence anisotropy to measure equilibrium and kinetic parameters—from picomolar kinase–substrate pairs to micromolar co-chaperone assemblies [67,68].
5. Experimental Methods for Detecting Protein–Protein Interactions
5.1 Overview and Classification
Methods for the experimental detection of PPIs are broadly categorized as in vitro, in vivo, and in silico approaches, each with characteristic strengths and limitations [69]. No single method is sufficient for comprehensive interactome mapping; convergent evidence from multiple orthogonal approaches is necessary to establish high-confidence interaction assignments [70].
5.2 In Vitro Methods
Co-immunoprecipitation (Co-IP) and affinity purification–mass spectrometry (AP-MS) represent the workhorses of large-scale PPI discovery [71]. Advances including SAINT and CompPASS scoring algorithms have substantially improved discrimination of specific interactions from non-specific binders [72]. Tandem affinity purification (TAP) employs a dual-tag system for two sequential purification steps under mild conditions, markedly reducing non-specific co-purifying proteins [73].
Proximity labeling approaches—BioID and TurboID—exploit promiscuous biotin ligases fused to bait proteins to biotinylate proximal proteins in living cells, enabling capture of transient and compartment-specific interactions typically disrupted during conventional co-IP [74,75]. Biophysical methods including surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and microscale thermophoresis (MST) provide quantitative kinetic and thermodynamic parameters essential for mechanistic understanding and drug discovery [76]. X-ray crystallography and NMR spectroscopy provide atomic-resolution structural information about complex architectures and conformational changes [77,78].
5.3 In Vivo Methods
The yeast two-hybrid (Y2H) system remains one of the most widely employed methods for large-scale PPI mapping [79]. In Y2H, a bait protein fused to a DNA-binding domain and a prey protein fused to a transcriptional activation domain reconstitute a functional transcription factor upon PPI, activating reporter genes [80]. Advances include matrix-based assays for systematic pairwise testing, improved vector systems, and use of multiple reporters with graduated sensitivity [81].
Limitations of Y2H include requirements for nuclear localization and susceptibility to auto-activation. Split-ubiquitin Y2H addresses the nuclear localization constraint for membrane proteins, while mammalian two-hybrid systems extend the approach to human cell contexts [82]. Fluorescence-based interaction assays in living cells—including FRET, BRET, and BiFC—permit real-time visualization of PPI dynamics in defined subcellular compartments [83,84].
5.4 Computational and In Silico Methods
In silico PPI prediction methods exploit protein sequences, three-dimensional structures, evolutionary conservation patterns, gene expression profiles, and phylogenetic distributions [85]. Sequence-based methods encompass correlated mutation analysis, interolog transfer, and machine learning classifiers trained on experimentally validated datasets [86]. Structure-based prediction exploits homologous binding modes, and docking algorithms including HADDOCK, ClusPro, Rosetta, and ZDock optimize surface complementarity and electrostatic potentials to generate PPI structural models [87,88].
Deep learning has recently transformed in silico PPI prediction. AlphaFold-Multimer demonstrates impressive accuracy in predicting heterodimer and homooligomer structures [89]. Universal In Silico Predictor of Protein–Protein Interactions (UNISPPI) and related tools provide binary interaction prediction from sequence information alone, enabling rapid pre-screening of large protein datasets [90].
6. Protein–Protein Interaction Networks: Organization and Analysis
6.1 Network Topology and Graph-Theoretic Properties
PPI networks are most naturally represented as undirected graphs in which nodes correspond to proteins and edges represent physical interactions [85]. Genome-wide interactome networks exhibit non-random topological properties: a scale-free degree distribution (hub proteins maintaining vastly more interactions than average), small-world characteristics (short average path lengths despite large network size), and modular organization [91,92]. Hub proteins are frequently essential for viability and often correspond to scaffolding proteins, chaperones, or proteins with multiple modular interaction domains [93].
Two categories of hubs are distinguished: date hubs, which interact with different partners in different cellular contexts, and party hubs, which engage multiple partners simultaneously within stable complexes [94]. Network analysis algorithms including k-clique community detection, Markov clustering, and spectral graph partitioning identify functionally coherent protein modules, and cross-species conservation of modules provides evidence for their functional importance [85,95].
6.2 Interactome Databases and Data Quality
Multiple curated databases aggregate experimentally validated PPI data, including BioGRID, the STRING database, IntAct, MINT, and the Human Reference Protein Interactome Mapping Project (HuRI) [96,97]. Standardization through the Proteomics Standards Initiative Molecular Interaction (PSI-MI) format has improved interoperability between databases [98]. Data quality remains a central challenge: high-throughput PPI datasets characteristically contain both false positives and false negatives, with overlap between datasets from different methods typically below 10% [99,100].
7. Protein–Protein Interactions in Signal Transduction Pathways
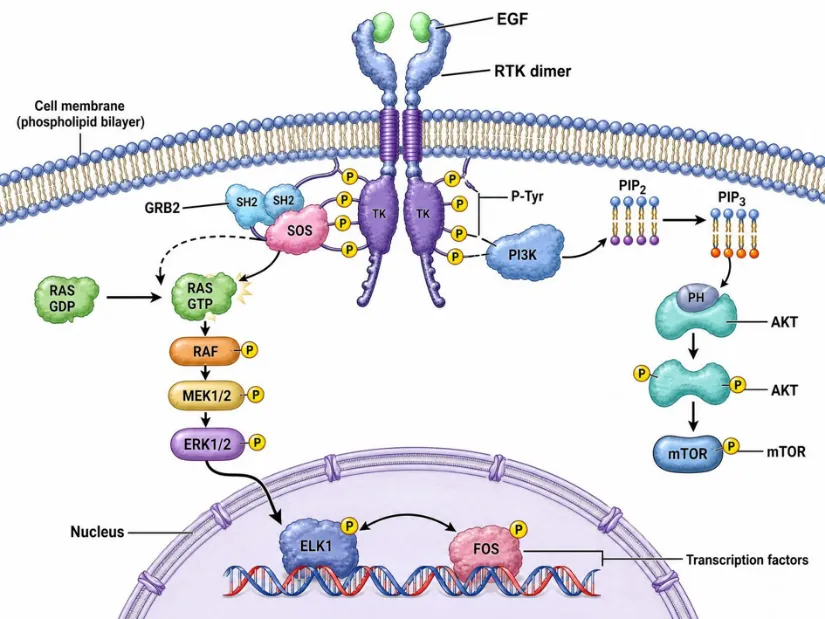
Figure 2. Receptor tyrosine kinase (RTK) signal transduction cascade illustrating sequential PPIs. EGF binding induces RTK dimerization and autophosphorylation of cytoplasmic tyrosines (pY). The GRB2 SH2 domain recognizes pY motifs and recruits the SOS guanine-nucleotide exchange factor, converting RAS-GDP to RAS-GTP. Active RAS engages the RAF→MEK→ERK kinase cascade, culminating in transcription factor nuclear translocation.
7.1 Architecture of Signaling Networks
Signal transduction pathways represent the cellular infrastructure through which extracellular information is sensed, processed, amplified, and translated into transcriptional, metabolic, and behavioral responses [1,3]. The molecular logic of signaling is fundamentally implemented through sequential PPIs: receptor activation triggers recruitment of adaptor proteins, activation of kinases, propagation through second messenger systems, and ultimately modulation of transcription factor activity [24,25].
The modular nature of signaling proteins is a central organizational principle. Signaling proteins characteristically contain multiple protein interaction domains—including SH2, SH3, PH, PDZ, and WD40 domains—that mediate selective recruitment of downstream effectors in a phosphorylation-dependent or lipid-dependent manner [19,20]. This modularity allows signal integration, signal diversification, and pathway insulation through scaffold proteins. Post-translational modification by kinases and phosphatases is the principal mechanism for dynamically regulating PPIs in signaling networks [24,25].
7.2 Crosstalk and Network Dynamics
Signaling networks are not linear cascades but extensively interconnected webs characterized by crosstalk—the regulatory influence of one pathway on another [23]. Crosstalk mechanisms include shared components, direct pathway-to-pathway interactions, transcriptional feedback loops, and competition for shared scaffolds or adaptors. Mathematical modeling approaches including ordinary differential equations (ODEs), Boolean network models, and agent-based simulations have been employed to understand emergent signaling properties such as bistability, oscillations, and signal amplification [10,30].
8. Protein–Protein Interactions in Metabolic Pathways and Gene Regulation
8.1 Metabolic Complexes and Metabolon Formation
Metabolic pathways are organized as supramolecular assemblies—termed metabolons—in which physically interacting enzymes channel substrates directly between active sites without equilibrating with the bulk cytoplasm [37]. The pyruvate dehydrogenase complex, citric acid cycle enzyme assemblies, and fatty acid synthase multienzyme complex are among the best-characterized examples of metabolically significant PPIs [28]. Metabolon formation offers kinetic advantages including substrate channeling, dynamic regulation, and co-localization of metabolic capacity to subcellular domains of highest demand. Flux balance analysis of large-scale metabolic networks complements the structural perspective provided by metabolon characterization [10].
8.2 Transcriptional Regulatory Networks and PPIs
Gene regulatory networks are fundamentally implemented through PPIs between transcription factors, co-activators, co-repressors, chromatin remodeling complexes, and the general transcription machinery [23]. Transcription factors function as dimers or higher-order assemblies whose partner selection determines DNA binding specificity and transcriptional outcome. The Mediator complex, a 26-subunit assembly bridging transcription factors and RNA polymerase II, illustrates the complexity of transcriptional PPI networks [20].
Chromatin-modifying complexes—including the SWI/SNF remodeling complex, Polycomb Repressive Complexes 1 and 2, and histone acetyltransferase complexes—constitute another major arena of PPI-mediated gene regulation. These assemblies are dynamically recruited to chromatin through interactions with modified histone marks, sequence-specific DNA binding factors, and regulatory non-coding RNAs [24,25].
9. Structural Protein Complexes: Ribosomes and the Cytoskeleton
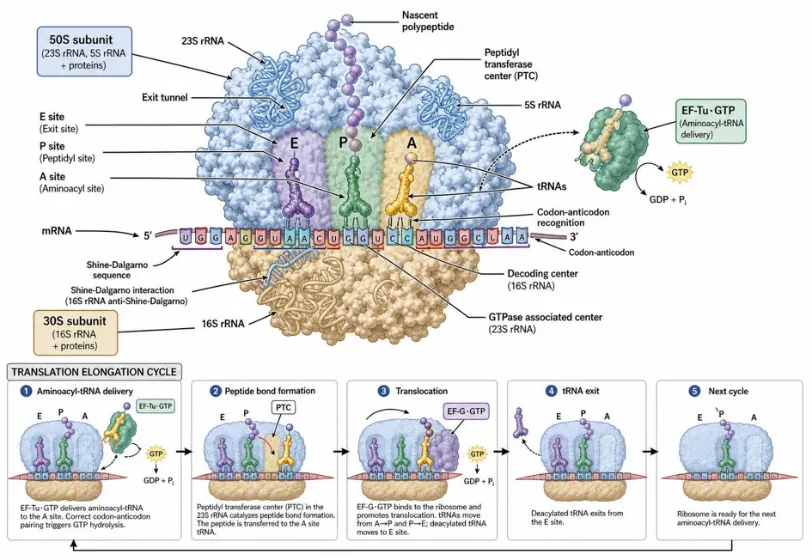
Figure 3. Architecture and elongation cycle of the 70S ribosome. The 50S large subunit (blue) and 30S small subunit (tan) form the A-, P-, and E-tRNA binding sites. EF-Tu·GTP delivers aminoacyl-tRNA to the A-site; peptide bond formation occurs at the peptidyl transferase center; EF-G·GTP catalyzes translocation, advancing mRNA by one codon.
9.1 Ribosomal Architecture and Translational PPIs
The ribosome is among the most intricate and evolutionarily conserved macromolecular machines in biology, consisting in prokaryotes of approximately 54 proteins and three ribosomal RNA molecules organized into two asymmetric subunits [11,12]. The large subunit catalyzes peptide bond formation at its peptidyl transferase center, while the small subunit performs mRNA codon decoding through interactions with aminoacyl-tRNAs. The elongation cycle—encompassing aminoacyl-tRNA delivery, peptidyl transfer, and translocation catalyzed by EF-G—is driven by transient PPIs that undergo dramatic conformational rearrangements coupled to GTP hydrolysis [11,21].
Ribosome-associated protein quality control (RQC) and ribosome rescue pathways involve specialized protein complexes that recognize stalled ribosomes and either resolve the stall or initiate targeted degradation of nascent polypeptides. Cryo-EM studies of translational machinery complexes have illuminated how these surveillance factors distinguish productive from aberrant translational intermediates [17,18].
9.2 Cytoskeletal Protein Networks
The cytoskeleton—comprising actin filaments, microtubules, intermediate filaments, and spectrin networks—is itself a vast PPI network in which structural proteins are regulated by an array of interacting factors including capping proteins, severing factors, polymerization nucleators, motor proteins, and crosslinkers [12]. The LINC (Linker of Nucleoskeleton and Cytoskeleton) complex, comprising SUN-domain proteins and KASH-domain nesprins, bridges the cytoplasmic cytoskeleton to the nucleoskeleton, physically coupling mechanical signals from the extracellular matrix to nuclear architecture and chromatin organization [12,23]. Disruption of LINC complex PPIs contributes to laminopathies, muscular dystrophies, and altered mechanosensing in cancer.
10. The Interactome Concept and Plant Protein Interaction Networks
The interactome—the complete network of physical protein interactions in a given organism or cellular context—represents the structural substrate of cellular function [5,6]. Comprehensive interactome mapping projects have been completed or are in progress for model organisms including Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and Homo sapiens, providing reference networks for comparative and translational analyses [69,70].
Plant interactomes have gained substantial attention with the availability of high-quality Arabidopsis thaliana and rice genome sequences [14]. The Arabidopsis Interactome Mapping Consortium generated a binary interaction map of approximately 6,200 interactions among 2,700 proteins using Y2H approaches, providing the first systematic framework for understanding protein connectivity in plant biology [14,79]. Clone-based proteomics approaches, facilitated by large-scale ORF collections and recombinational cloning technologies, have enabled systematic interaction mapping in plants. Split-ubiquitin assays extend interaction mapping to membrane proteins, a functionally important but technically challenging class of plant interactome components [82,84].
11. PPIs in Disease: Oncology, Neurodegeneration, and Proteostasis
11.1 Oncological Roles of Pathological PPIs
Cancer is fundamentally a disease of deregulated cellular signaling, and aberrant PPIs constitute central molecular pathogenic mechanisms [15,16]. The MDM2–p53 interaction, through which the MDM2 E3 ubiquitin ligase targets p53 for proteasomal degradation, is hyperactivated in many tumors retaining wild-type p53 [17]. The BCL-2 family of anti-apoptotic proteins engages pro-apoptotic partners BIM, BAX, and BAK through BH3 domain interactions; overexpression of BCL-2, BCL-XL, or MCL-1 suppresses apoptosis and promotes chemoresistance [18]. Oncogenic transcription factors—including MYC, β-catenin/TCF, STAT3, and androgen/estrogen receptors—drive malignancy through PPI networks activating proliferative gene expression programs [15,16].
11.2 Neurodegenerative disease and Aberrant PPI Assemblies
Neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) share a hallmark of aberrant protein aggregation, in which normally soluble proteins assemble into toxic oligomeric or fibrillar species through pathological PPIs [17]. In AD, amyloid-β (Aβ) peptides self-associate through β-sheet stacking interactions to form soluble oligomers and insoluble amyloid fibrils [17,18]. Tau protein in AD, α-synuclein in PD, huntingtin in HD, and mutant SOD1 in ALS undergo pathological PPIs producing conformationally distinct aggregate strains with different propagation properties and neurotoxicity profiles. Advances in cryo-EM have enabled near-atomic resolution determination of tau filament structures from patient brain tissue, revealing disease-specific folding topologies underlying clinicopathological diversity of tauopathies [13,14].
11.3 Proteostasis Networks and Chaperone PPIs
Proteostasis—the dynamic equilibrium of protein production, folding, trafficking, and degradation—is maintained by an extensive PPI network involving molecular chaperones, co-chaperones, ubiquitin–proteasome system components, and autophagy machinery [17]. The Hsp70/Hsp90 chaperone system engages clients through transient PPIs mediated by substrate-binding domains, stabilizing partially folded intermediates and preventing aberrant aggregation. Proteostatic collapse—characterized by accumulation of ubiquitinated inclusion bodies—is observed in aging and disease states. Therapeutic strategies targeting proteostasis PPIs include pharmacological chaperones, disaggregases, and proteolysis-targeting chimeras (PROTACs) that redirect E3 ubiquitin ligases toward disease proteins [15,16].
12. Advances in Structural Biology: Artificial Intelligence and Cryo-Electron Microscop
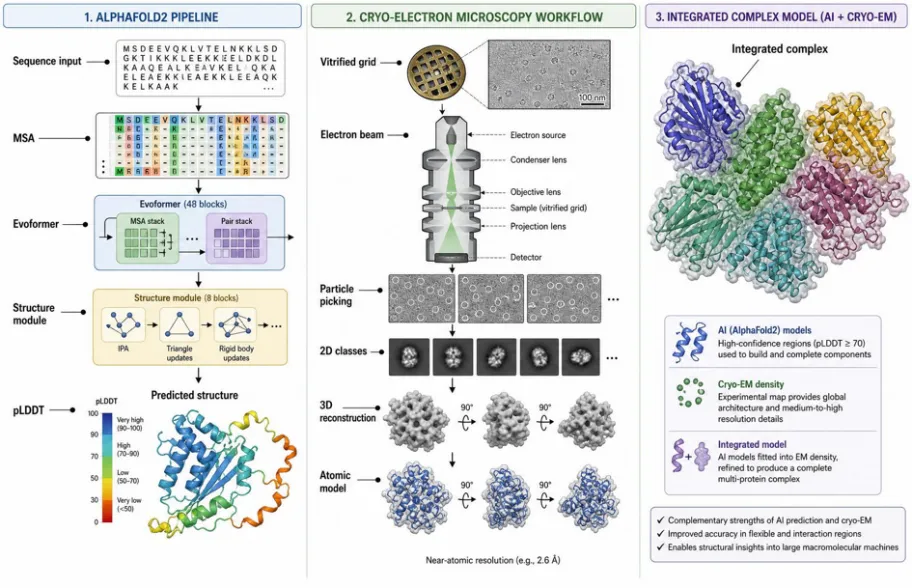
Figure 4. Integrated structural-biology workflow combining AI-driven prediction and cryo-EM. Left: AlphaFold2 derives multiple sequence alignments (MSA), processes them through the Evoformer and Structure module, and yields a predicted multimeric complex with per-residue pLDDT confidence. Right: cryo-EM vitrifies the specimen, collects movies on a direct electron detector, generates 2D class averages, and reconstructs a 3D density map (\~3 Å) for atomic-model fitting.
12.1 AI-Driven Protein Structure Prediction
The protein folding problem has been fundamentally transformed by deep learning [13,19]. AlphaFold2, developed by DeepMind, achieves near-experimental accuracy in structure prediction by integrating evolutionary information (multiple sequence alignments), pairwise residue distance matrices, and attention-based neural networks that iteratively refine structural predictions [13]. Performance in the CASP14 benchmarks demonstrated AlphaFold2's superiority over all prior methods across most target categories, with median TM-scores exceeding 0.90 for many domain types [13].
RoseTTAFold and ESMFold represent complementary deep learning architectures achieving AlphaFold2-comparable accuracy for many protein families while offering computational advantages for specific applications. These tools have been deployed to generate structure predictions for essentially all known protein sequences in UniProt, creating an unprecedented structural proteomics resource [13,19]. AlphaFold-Multimer enables complex structure prediction with accuracy competitive with experimental methods for many heterodimeric and homo-oligomeric assemblies [89]. Generative AI models—including diffusion-based approaches such as RFdiffusion and ProteinMPNN—enable de novo protein design, extending AI applications from structural prediction to functional protein engineering [19].
12.2 Cryo-Electron Microscopy Revolution
Cryo-electron microscopy (cryo-EM), in which biological specimens are rapidly vitrified and imaged at cryogenic temperatures, has undergone a resolution revolution driven by direct electron detectors, improved phase contrast, and advanced image processing algorithms [13,20]. These advances have enabled routine determination of macromolecular structures at resolutions below 3 Å without requiring crystallization—a major bottleneck for large, flexible, and membrane-associated complexes [20].
Cryo-EM has proven transformative for characterizing large multiprotein assemblies including ribosomes, proteasomes, spliceosomes, CRISPR-Cas surveillance complexes, and GPCRs. Time-resolved cryo-EM approaches now enable visualization of dynamic conformational transitions within functional cycles [20]. Application of cryo-EM to disease-associated protein aggregates has revealed tau filament structures from Alzheimer's disease brain tissue with disease-specific conformations not predictable from isolated tau fragments, directly connecting structural polymorphisms to clinical phenotypes [17,20].
13. Therapeutic Targeting of Protein–Protein Interactions
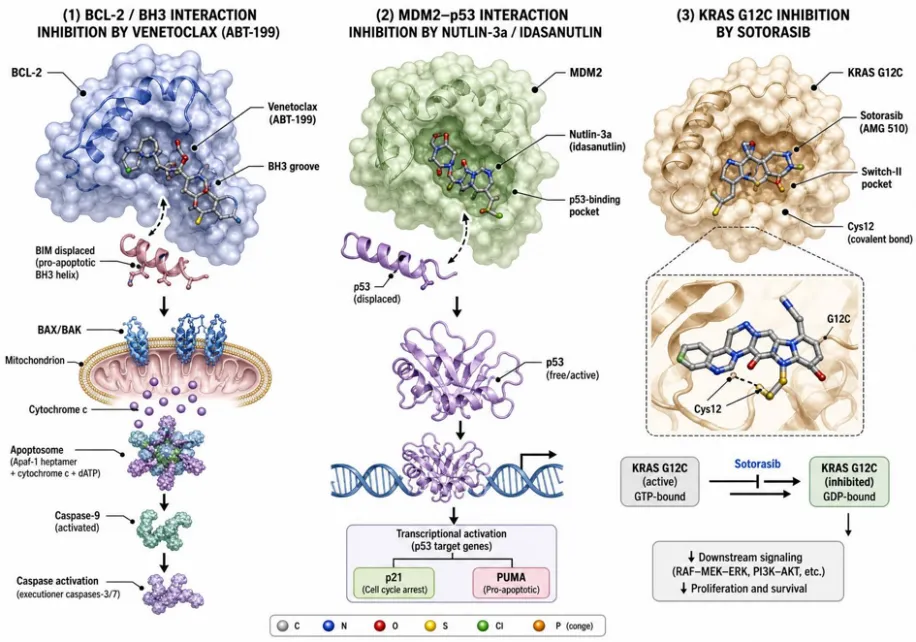
Figure 5. Small-molecule inhibition of disease-relevant protein–protein interactions. (A) MDM2 (grey) bound to the p53 transactivation-domain helix (red); a Nutlin-class small-molecule inhibitor (green sticks) occupies the Phe19/Trp23/Leu26 hot-spot pocket, displacing p53 and restoring tumor-suppressor activity. (B) BCL-2 (blue) engaging the BH3 helix of BIM (orange) through the P1–P4 hydrophobic groove; Venetoclax (ABT-199) competes for the groove, releasing pro-apoptotic effectors in BCL-2-dependent malignancies.
13.1 Rationale and Challenges
PPIs, with their large, relatively flat contact surfaces (typically 1,500–3,000 Ų), were long considered intractable for small-molecule intervention [15,16]. However, structural analyses reveal that interface surfaces contain pockets, grooves, and hot spot clusters accommodating small molecules [42,43]. The recognition that hot spot residues contribute disproportionately to binding energy implies that small molecules occupying these hot spots need not replicate the full interface but merely mimic its most energetically critical elements [44,45]. This hot spot-centric drug design paradigm has inspired fragment-based drug discovery campaigns targeting PPI interfaces [38,39].
13.2 Classes of PPI Modulators
PPI modulators fall into three primary classes: (i) monoclonal antibodies, (ii) peptides and peptidomimetics, and (iii) small-molecule inhibitors or stabilizers [15,16]. Monoclonal antibodies achieve exquisite target specificity and high affinity but are limited by poor cell permeability, inability to cross the blood–brain barrier, and high manufacturing costs. Peptides derived from PPI interface sequences provide structural mimics of the natural binding epitope. Chemical strategies including peptide stapling, hydrogen-bond surrogates, and N-methylation improve metabolic stability and cellular uptake [18,21].
Small-molecule PPI inhibitors and stabilizers represent the most pharmacologically versatile class of modulators. Venetoclax (ABT-199), a BH3 mimetic targeting BCL-2, received FDA approval for chronic lymphocytic leukemia and represents the first approved small-molecule PPI inhibitor [17,18]. MDM2 inhibitors are in clinical trials for solid tumors, while STAT3, IAP, and RAS–SOS inhibitors are in various stages of development [15,16]. Importantly, a fourth class—PPI stabilizers—has gained traction. Rapamycin stabilizes the FKBP12–mTOR complex, and lenalidomide and related IMiDs induce neo-PPIs between the cereblon E3 ligase and neosubstrates, effectively reprogramming ubiquitin ligase specificity [21].
13.3 Strategies for PPI Drug Discovery
The discovery of PPI modulators employs complementary strategies including high-throughput screening (HTS) of diverse compound libraries, fragment-based drug discovery (FBDD), computational virtual screening, and structure-based design [15,18]. Fragment-based approaches have proven particularly successful for PPI targets, as small fragments (150–300 Da) can occupy hot spots with measurable affinity (typically mM to μM Kd) elaborated through medicinal chemistry. Phage display, mRNA display, and ribosome display enable selection of peptidic binders against PPI interfaces with high efficiency and nanomolar affinity [21].
CONCLUSION AND FUTURE PERSPECTIVES
Protein–protein interactions constitute the molecular foundation of all cellular functions, from the most fundamental biochemical transformations to the most complex systems-level biological responses. The past decade has witnessed remarkable progress in our ability to map, characterize, model, and therapeutically exploit these interactions. High-throughput interactomics, structural proteomics, network biology, and clinical PPI inhibitor development have converged to create a transformative era in molecular biology and medicine.
Looking forward, the integration of AI-driven structure prediction with experimental structural biology promises to generate increasingly comprehensive and accurate structural interactomes. AlphaFold-Multimer and related tools will drive systematic structural annotation of known interactions and provide candidate structural models for interactions lacking experimental structural data. The development of PPI-targeting therapeutics will be accelerated by improved understanding of interface druggability, advances in intracellular delivery of peptidic modulators, and the emerging PROTAC/molecular glue paradigm for targeting previously undruggable proteins.
Single-cell and spatial proteomics technologies will enable mapping of cell-type-specific and spatiotemporally resolved PPI networks, revealing how interaction dynamics vary across cell states, developmental transitions, and disease progression. Advances in cryo-electron tomography will enable in situ structural determination of complexes in their native cellular environment. The growing appreciation of disordered proteins, weak transient interactions, and condensate-mediated compartmentalization as drivers of PPI networks opens new avenues for basic discovery and therapeutic intervention. The study of protein–protein interactions remains one of the most dynamic and consequential frontiers in the life sciences.
REFERENCES



Afreen Katharbasha, Pratiksha Jadhao, Shivprasad Dhage, Protein–Protein Interactions in Cellular Machinery: Molecular Mechanisms, Functional Networks, and Therapeutic Opportunities, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 950-969. https://doi.org/10.5281/zenodo.21194044
 10.5281/zenodo.21194044
10.5281/zenodo.21194044