
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Vidya Niketan Institute of Pharmacy and Research Center, Bota.
Drug design is a multidisciplinary endeavor focused on the discovery and development of novel therapeutic agents with optimized efficacy, safety, and pharmacokinetic properties. It synthesizes principles from medicinal chemistry, pharmacology, molecular biology, and computational modeling to identify and refine lead compounds. Modern approaches include structure-based drug design (SBDD), which leverages the three-dimensional structures of biological targets, and ligand-based drug design (LBDD), which utilizes the chemical features of known active compounds. Recent advances in computational methods, high-throughput screening, and artificial intelligence have significantly accelerated the processes of lead identification and optimization. Iterative refinement is employed to improve properties such as binding affinity, selectivity, solubility, and metabolic stability, while minimizing off-target effects and toxicity. Ultimately, drug design plays a critical role in bridging the gap between target identification and clinical application, driving the development of innovative therapeutics for a wide range of diseases.
Drug design is a systematic and strategic process aimed at discovering and developing new pharmaceutical agents that can precisely interact with specific biological targets to treat or prevent diseases. It acts as a vital bridge between fundamental biomedical research and clinical application, with the goal of producing molecules that exhibit high specificity and potency, along with favorable pharmacokinetic properties and safety profiles.
The field primarily involves two main approaches: structure-based drug design (SBDD), which utilizes the three-dimensional structure of the target biomolecule to guide compound design, and ligand-based drug design (LBDD), which relies on knowledge of existing biologically active molecules to create new candidates. Advances in computational chemistry, molecular modelling, and bioinformatics have greatly improved the efficiency and accuracy of both methods.[1] Modern drug design integrates in silico techniques such as molecular docking, quantitative structure–activity relationship (QSAR) modelling, and pharmacophore mapping to predict interactions between molecules and their targets. These methods, combined with high-throughput screening and combinatorial chemistry, have accelerated the discovery process, reduced costs, and increased the likelihood of identifying successful drug candidates. Overall, drug design represents the convergence of chemistry, biology, and computational science, driving innovation in the creation of targeted therapeutics for a wide range of health conditions.[2]. Drug design is a multidisciplinary process aimed at identifying and developing new therapeutic agents that can specifically interact with biological targets to treat, manage, or prevent diseases. It represents a crucial stage in the drug discovery pipeline, transforming basic biological and chemical knowledge into practical medicinal solutions. The process seeks to create molecules that possess optimal binding affinity, high selectivity, minimal toxicity, and favourable pharmacokinetic properties such as absorption, distribution, metabolism, and excretion (ADME).[3]. In recent decades, technological advancements have significantly transformed the field. Computational tools now play a central role, enabling in silico screening of vast chemical libraries, prediction of molecular interactions, and optimization of lead compounds through molecular docking, pharmacophore modelling, and quantitative structure–activity relationship (QSAR) analysis. These approaches, combined with high-throughput screening, combinatorial chemistry, and artificial intelligence, have accelerated the pace of discovery, reduced research costs, and increased the probability of clinical success.[4]. Drug design also involves iterative refinement, where candidate molecules are continuously modified to enhance desired properties and eliminate drawbacks. This process requires close integration between medicinal chemistry, pharmacology, toxicology, and formulation science. Ultimately, successful drug design not only contributes to medical innovation but also addresses unmet healthcare needs, paving the way for the development of next-generation therapeutics targeting complex and challenging diseases.[5]
History and Evolution of Drug Design
The history of drug design can be traced back to ancient civilizations, where medicinal preparations were derived directly from natural sources such as plants, minerals, and animal products. Early drug discovery was based largely on trial-and-error methods, traditional knowledge, and empirical observations rather than a scientific understanding of disease mechanisms. Herbal remedies, alkaloids, and crude extracts formed the basis of ancient pharmacotherapy. The 19th century marked the beginning of the scientific era of drug discovery. The isolation of pure compounds, such as morphine from opium and quinine from cinchona bark, allowed for the precise study of chemical structures and pharmacological effects. The development of organic chemistry during this period enabled scientists to synthesize new molecules and modify existing natural compounds for improved therapeutic properties.[6] In the early 20th century, the concept of “magic bullets” proposed by Paul Ehrlich revolutionized drug development. Ehrlich envisioned designing chemicals that could selectively target disease-causing agents without harming healthy tissues, laying the foundation for targeted drug design. This era saw the discovery of sulphonamides and the introduction of antibiotics like penicillin, which transformed medicine by providing effective treatments for infectious diseases. The mid-20th century brought the rise of rational drug design, supported by advancements in biochemistry and molecular biology. Scientists began to understand enzymes, receptors, and other biomolecules as drug targets. The discovery of DNA’s structure in 1953 and the growth of molecular pharmacology paved the way for drugs designed to interact with specific biological pathways.[7]. From the late 20th century onwards, the integration of computational methods revolutionized drug design. Structure-based drug design (SBDD) emerged as a powerful tool, using three-dimensional structural information obtained from X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy. Ligand-based drug design (LBDD) also advanced, relying on data from known active compounds to design analogs with improved properties. In the 21st century, drug design has entered an era of precision and personalization. Computational chemistry, molecular docking, high-throughput screening, and artificial intelligence are now integral parts of the process. These technologies allow rapid screening of millions of compounds, prediction of pharmacokinetic and toxicity profiles, and optimization of drug candidates before synthesis. Additionally, advances in genomics and proteomics have paved the way for personalized medicine, where therapies can be tailored to an individual’s genetic makeup and disease profile.[8]
Introduction to CADD
Computer-Aided Drug Design (CADD) is a modern approach in pharmaceutical research that utilizes computational tools and modelling techniques to identify, design, and optimize potential drug candidates. It plays a crucial role in reducing the time, cost, and resources required for traditional drug discovery by allowing scientists to simulate and analyze molecular interactions before laboratory synthesis and testing.[9] CADD integrates principles from medicinal chemistry, molecular biology, bioinformatics, and structural biology to predict how a compound will interact with a specific biological target. By employing mathematical models, molecular docking, virtual screening, and quantitative structure–activity relationship (QSAR) analyses, researchers can assess the binding affinity, selectivity, and pharmacokinetic properties of compounds in silico.
There are two main categories of CADD:
Chemical Structure Representation
Chemical structure representation is the method of illustrating the arrangement of atoms in a molecule and the chemical bonds between them. It serves as a visual and logical way to understand a compound’s composition, geometry, and reactivity. Representations can be two-dimensional (2D) for structural clarity or three-dimensional (3D) for spatial and conformational analysis.
1. Types of Chemical Structure Representation
a) Molecular Formula
b) Lewis Structure
c) Structural Formula (2D Representation)
d) Skeletal (Line-Angle) Formula
e) Ball-and-Stick Model (3D Representation)
f) Space-Filling Model
g) Computer-Aided Models (CADD)
2. Importance in Drug Design and CADD
Chemical Database Search
A chemical database search is the process of retrieving information about chemical compounds, their properties, and related data from specialized digital repositories. These databases are essential tools in drug discovery, chemical research, and CADD (Computer-Aided Drug Design), enabling scientists to identify known compounds, predict properties, and explore potential new molecules.
1. Types of Chemical Databases
a) Structure Databases
Contain detailed molecular structures with bonding information.
Examples:
b) Spectral Databases
Store experimental spectra such as IR, NMR, MS.
Examples:
c) Bioactivity Databases
Provide data on biological activity of compounds against various targets.
Examples:
d) Commercial Databases
Require paid access; often have advanced search tools and proprietary data.
Examples:
2. Search Methods in Chemical Databases
3. Applications in Drug Design
Nice — I’ll unpack each pharmacophore element and then explain how models are built, represented, validated, and used in practice. I’ll keep it practical (what it means physically, how it’s encoded in software, and what to watch out for).[3]
Key pharmacophore features — detailed
Hydrogen-bond donors (HBD)
Hydrogen-bond acceptors (HBA)
Aromatic/π systems
Positive / Negative ionizable groups (charged centres)
Metal-binding features
Halogen-bonding / special interactions (optional advanced features)
Excluded volumes (steric constraints)
Types of pharmacophore models — how they differ
Ligand-based pharmacophores
Structure-based pharmacophores
Ensemble / hybrid pharmacophores
Typical workflow to create and use a pharmacophore model
Validation & performance metrics (practical)
Practical tips & common pitfalls
Common tools (short list)
Docking
Docking is a computational technique used in Computer-Aided Drug Design (CADD) to predict how a small molecule (ligand) binds to a specific site on a macromolecular target (such as a protein or enzyme). Its goal is to determine the optimal binding orientation and estimate the strength of the interaction between the ligand and the target, which can help identify and design new drug candidates.
1. Purpose of Docking
2. Types of Docking
a) Rigid Docking
b) Flexible Docking
3. Steps in Docking Process
4. Common Docking Tools
5. Applications in Drug Design
6. Advantages
7. Limitations
ADMET
ADMET stands for Absorption, Distribution, Metabolism, Excretion, and Toxicity — the key pharmacokinetic and safety properties that determine whether a drug candidate will work in the body as well as it does in the computer. It’s often evaluated in silico early in the design process to save time, money, and avoid failures in later animal/human studies.
1. Absorption (A)
2. Distribution (D)
3. Metabolism (M)
4. Excretion (E)
5. Toxicity (T)
Typical In-Silico ADMET Workflow
Why ADMET is Critical
Sources of Natural Drugs
a. Plant-derived drugs
b. Microbial-derived drugs
c. Marine-derived drugs
d. Animal-derived drugs
3. Advantages of Natural Drug Sources[6]
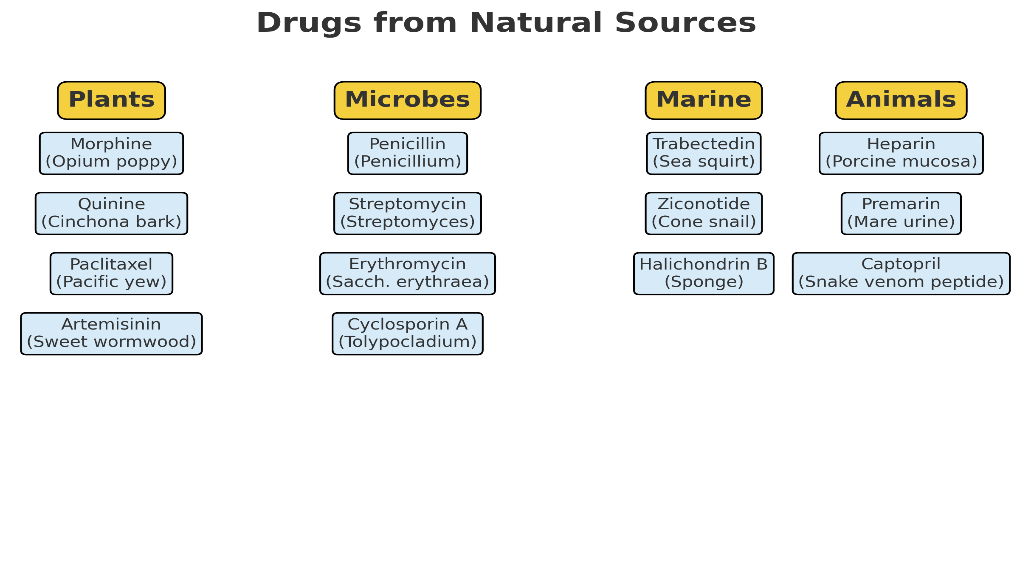
Fig 1. Sources of natural drugs
CONCLUSION
Drugs derived from natural sources have been instrumental in shaping modern medicine, offering an exceptional range of chemical structures and unique mechanisms of action that have inspired numerous therapeutic agents. Natural products from plants, microbes, marine organisms, and animals have served not only as direct remedies but also as valuable lead compounds for synthetic modification and optimization. Despite significant advances in synthetic chemistry and biotechnology, natural sources remain an irreplaceable reservoir for drug discovery due to their structural diversity, potent biological activity, and evolutionary refinement. The fusion of traditional knowledge with modern analytical techniques, high-throughput screening, and computational tools continues to unlock new possibilities, reinforcing the relevance of natural products in combating emerging and drug-resistant diseases. By preserving biodiversity and exploring under-researched ecosystems, the potential for discovering innovative treatments can be greatly expanded; ensuring natural drug discovery remains a vital pillar of future healthcare.
REFERENCE


Vanita Katore, Tanishk Kardile*, Shubham Gawali, Review on Drug Design, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 9, 3094-3107 https://doi.org/10.5281/zenodo.17204936
 10.5281/zenodo.17204936
10.5281/zenodo.17204936