
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacology, Channabasweshwar Pharmacy College (Degree), Latur
The rising global burden of diabetes mellitus underscores the urgent need for innovative and effective therapeutic interventions. Central to this process is the development and application of reliable experimental models for preclinical screening of antidiabetic compounds. This review offers a comprehensive overview of both traditional and advanced in vitro and in vivo models used in antidiabetic drug discovery. In vitro assays-including enzyme inhibition, glucose uptake, insulin secretion, and 3D cell cultures-facilitate high-throughput, mechanistic evaluation of candidate compounds. Emerging technologies such as organ-on-a-chip platforms and iPSC-derived ?-cells have further enhanced the physiological relevance and predictive value of early-stage screening. Complementing these are in vivo models, ranging from chemically or genetically induced diabetic rodents to humanized and CRISPR/Cas9-edited animals, which allow for systemic evaluation of pharmacodynamics, toxicity, and inter-organ metabolic interactions. Zebrafish and multi-organ microfluidic systems also present powerful, scalable tools for compound screening and mechanistic insights. By systematically comparing established and novel experimental models, this review highlights current capabilities and limitations while proposing an integrative screening strategy to accelerate the development of next-generation antidiabetic therapies.
"Diabetes mellitus is a chronic metabolic disorder characterized by elevated levels of glucose in the blood due to the body's inability to produce enough insulin or effectively use the insulin it produces. This condition affects how the body converts food into energy and, if left unmanaged, can lead to serious complications involving the heart, kidneys, eyes, and nerves."[1] The global prevalence of diabetes has nearly doubled over the past three decades. In 2021, approximately 529 million people globally were living with diabetes, primarily type 2 [which accounted for about 96% of cases] This trend is expected to accelerate, with more than 1.31 billion people projected to have diabetes by 2050. [2] Another authoritative data source, the IDF Diabetes Atlas 2025, reports that currently 589 million adults [ages 20–79] have diabetes-about 1 in 9 adults. This number is anticipated to rise to 853 million by 2050. Alarmingly, approximately 43% of adults with diabetes [around 252 million people] remain undiagnosed, with almost 90% of those undiagnosed living in low- and middle-income countries. [3]
Table 1. Global Diabetes Prevalence and Future Projections According to GBD [2021] and IDF Atlas [2025]
|
Data Source |
Key Figures |
|
GBD [2021] |
~529 million with diabetes; projected >1.31 billion by 2050; ~96% type 2 |
|
IDF Atlas [2025] |
589 million adults [20–79] currently with diabetes; projected 853 million by 2050; ~43% undiagnosed |
The two principal categories are Type 1 diabetes mellitus [T1DM]-an autoimmune destruction of pancreatic β‑cells leading to absolute insulin deficiency-and Type 2 diabetes mellitus [T2DM] characterized by a combination of insulin resistance and relative insulin insufficiency. Gestational diabetes mellitus [GDM] arises during pregnancy and indicates an elevated future risk for T2DM. [4] Beyond these major types, there are "other specific types" caused by diverse etiologies such as monogenic defects [e.g., MODY], genetic syndromes, pancreatic injury [Type 3c], infections, endocrinopathies, or drug-induced mechanisms. This expanded classification underscores the clinical and pathophysiological complexity of diabetes, facilitating more accurate diagnosis and individualized treatment strategies. [5]
The development of effective antidiabetic agents fundamentally depends on robust preclinical screening models that reliably simulate both the physiological complexity of diabetes and the pharmacological effects of candidate compounds. In vitro assays-including α-glucosidase and α-amylase inhibition tests, glucose uptake assays, insulin secretion assays, and high-throughput cell‑based systems-are indispensable in early-stage drug discovery. These methods are cost-efficient, ethically favorable, and facilitate rapid screening to identify active compounds and explore mechanisms of action, offering clear advantages in throughput and control over experimental variables. [6] Nevertheless, vivo models-such as chemically induced, genetically modified, diet-induced, or transgenic animal models-are critical to assessing pharmacokinetics, systemic efficacy, toxicity, and complex multi-organ interactions, though they entail higher costs, ethical challenges, and interspecies translational limitations. [7] Comprehensive reviews emphasize that integrating in vitro screening with vivo validation enhances predictive reliability and accelerates progression from candidate identification to clinical potential. Therefore, a dual strategy combining high-throughput in vitro platforms with confirmatory in vivo models remains essential for the efficient and translational development of novel antidiabetic therapies. [8]
This review aims to provide a comprehensive evaluation of the currently established in vitro and in vivo screening models used in the discovery and development of antidiabetic drugs, highlighting their strengths and limitations. Furthermore, it explores novel and emerging experimental models that offer enhanced predictive value and translational relevance in antidiabetic drug screening. By systematically comparing traditional and innovative approaches, this review seeks to guide future research towards more efficient and reliable screening strategies in diabetes therapeutics development.
2. Experimental models for antidiabetic drug screening [6-10]
Table 2. Overview of Existing and Advanced In Vitro and In Vivo Diabetes Models.
|
Category |
Model Type |
Description |
Examples |
|
Existing [Traditional] Models |
In Vitro Models |
Biochemical and cell-based assays for enzyme inhibition, glucose uptake, and insulin secretion. |
α-glucosidase & α-amylase inhibition assays, glucose uptake in muscle/adipocyte cells [L6, 3T3-L1], insulin secretion in β-cell lines [INS-1, MIN6], cytotoxicity assays [MTT, LDH] |
|
In Vivo Models |
Animal models replicating diabetes via chemicals, genetic mutations, or diet-induced insulin resistance. |
Streptozotocin [STZ]-induced diabetes, Alloxan-induced diabetes, db/db mice, ob/ob mice, Zucker diabetic fatty [ZDF] rats, high-fat diet rodents, pancreatectomy models, Goto-Kakizaki [GK] rats |
|
|
New [Emerging/ Advanced] Models |
In Vitro / Ex Vivo |
Advanced culture systems providing more physiological relevance and complex interactions. |
3D pancreatic islet/organoid cultures, microfluidic “organ-on-a-chip” systems, human iPSC-derived β-cells, high-throughput screening platforms with multi-parametric readouts |
|
In Vivo Models |
Genetically engineered or alternative species models for improved human disease modeling and translational accuracy. |
CRISPR/Cas9 gene-edited mice targeting diabetes genes, zebrafish diabetes models, humanized mice with human cell grafts, multi-organ interaction animal models [liver-pancreas-muscle crosstalk] |
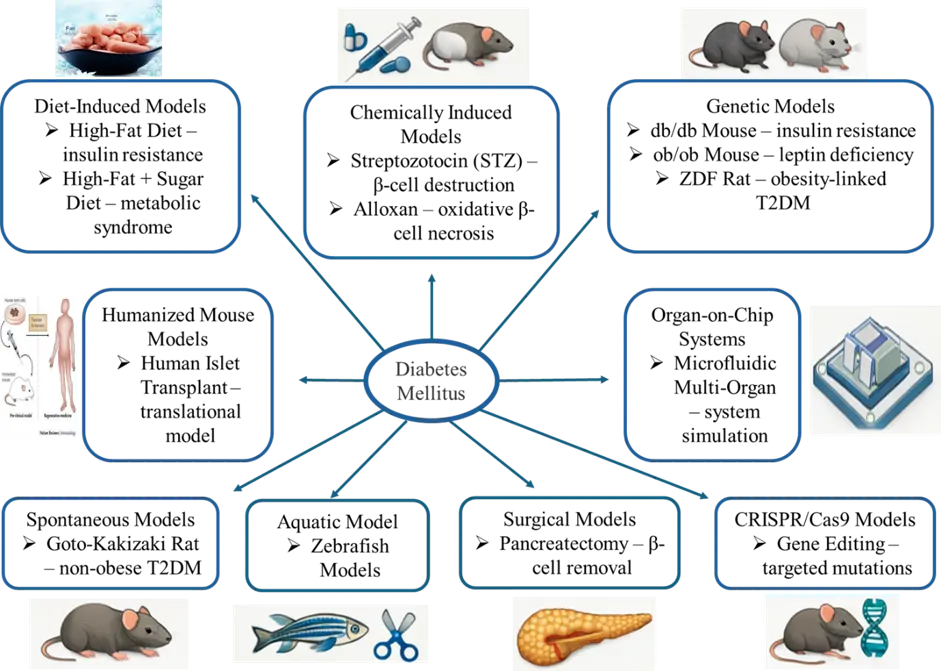
Figure 1. Overview of in vivo, in vitro, ex vivo, and emerging experimental models used in Diabetes Mellitus studies.
2.1 Cell-Based and Biochemical In Vitro Models in Antidiabetic Research
In vitro screening models are foundational to early-stage antidiabetic drug discovery, offering cost-effective, reproducible, and ethically favourable alternatives to in vivo systems. Among these, enzyme inhibition assays, particularly those targeting α-glucosidase and α-amylase, are widely utilized to identify agents capable of delaying carbohydrate digestion and attenuating postprandial glucose spikes-key therapeutic goals in type 2 diabetes mellitus [T2DM] management. [7] In addition to biochemical assays, cell-based models such as glucose uptake assays in 3T3-L1 adipocytes and L6 myotubes, or insulin secretion assays using β-cell lines like INS-1 and MIN6, provide functional insight into insulin sensitivity and pancreatic activity. [6] To enhance physiological relevance, emerging technologies such as three-dimensional [3D] cell cultures, microfluidic organ-on-a-chip platforms, and human iPSC-derived β-cells are increasingly integrated into drug screening pipelines. These systems better replicate the structural and functional microenvironment of human tissues and support dynamic, long-term assessments of drug efficacy and toxicity. [11,12] For instance, organ-on-chip models mimicking pancreatic-liver crosstalk allow for real-time monitoring of insulin response under flow conditions. [12] Additionally, microfluidic high-throughput screening platforms and multi-organ chip systems enable predictive modeling of systemic metabolic interactions, thereby narrowing the translational gap between in vitro data and clinical outcomes. [13,14]
2.1.1 Enzyme Inhibition Assays
The inhibition of carbohydrate-hydrolyzing enzymes, particularly α-amylase and α-glucosidase, is a widely studied therapeutic strategy for managing postprandial hyperglycaemia in individuals with type 2 diabetes mellitus. α-Amylase initiates the hydrolysis of dietary starch into smaller oligosaccharides, which are subsequently broken down into glucose by α-glucosidase in the small intestine [15]. Inhibiting these enzymes delays glucose release and absorption, thereby flattening post-meal blood glucose spikes [16]. Various in vitro assays have been established to evaluate the inhibitory activity of natural and synthetic compounds against these enzymes. Typically, α-amylase inhibition is measured using starch substrates and colorimetric detection of reducing sugars, while α-glucosidase inhibition is assessed using substrates like p-nitrophenyl-α-D-glucopyranoside [pNPG], where the release of p-nitrophenol is quantified spectrophotometrically [17]. Such assays are essential in the preliminary screening of bioactive compounds for potential antidiabetic properties and support the identification of lead candidates for further pharmacological development.
2.1.2 Glucose Uptake Assays
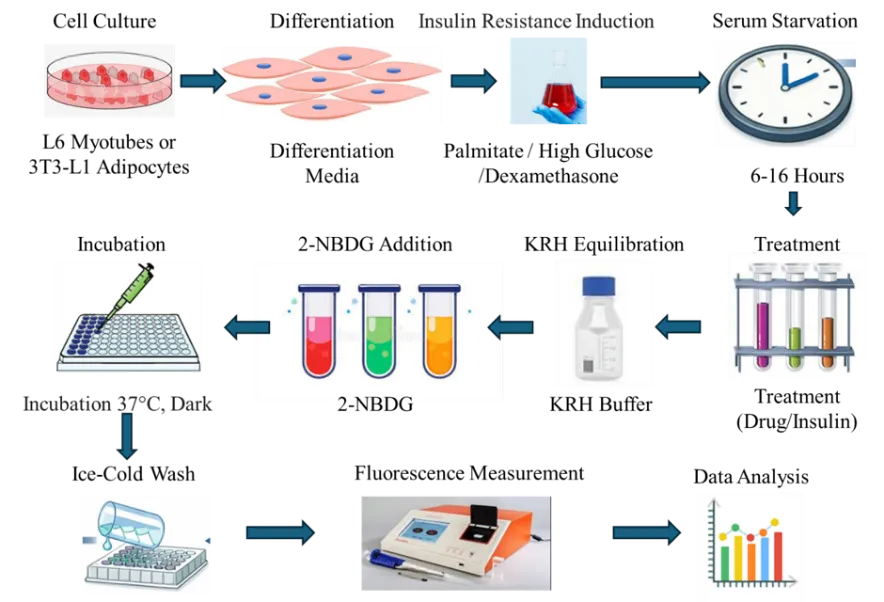
Figure 2. Schematic overview of the in vitro experimental protocol for insulin resistance induction and fluorescence-based glucose uptake analysis.
Glucose uptake assays are widely used to evaluate the insulin-mimetic or insulin-sensitizing effects of bioactive compounds in vitro. Commonly employed cell lines include L6 and C2C12 myotubes [derived from skeletal muscle] and 3T3-L1 adipocytes, both of which are metabolically active and responsive to insulin signalling [18]. These cells are differentiated under specific conditions to promote insulin-responsive glucose transporter [GLUT4] expression, which is critical for assessing glucose uptake activity. Typically, 2-deoxy-D-glucose or fluorescent glucose analogs such as 2-NBDG are used as tracers to quantify glucose uptake via spectrophotometric or fluorometric methods [19]. Compounds that enhance glucose uptake in these models may improve insulin sensitivity or mimic insulin action, making them promising candidates for antidiabetic drug development. The stimulation of glucose uptake in muscle and adipose tissues is particularly significant, as these are the primary sites of insulin-mediated glucose disposal in vivo [18]. Thus, these assays serve as a vital tool in early-stage screening for potential therapeutic agents targeting type 2 diabetes mellitus.
2.1.3 Insulin Secretion Assays
Insulin secretion assays using pancreatic β-cell lines, such as INS-1 and MIN6, are widely employed to evaluate the insulinotropic potential of natural or synthetic compounds. These cell lines retain many physiological characteristics of native pancreatic β-cells, including the ability to secrete insulin in response to elevated glucose levels [20]. Glucose-stimulated insulin secretion [GSIS] is typically measured under basal and high-glucose conditions, often in the presence or absence of test compounds, to determine their effect on insulin release. The insulin content in the culture supernatant is quantified using enzyme-linked immunosorbent assay [ELISA] or radioimmunoassay [RIA] techniques [21]. Enhancing GSIS is a critical therapeutic strategy in type 2 diabetes, where β-cell dysfunction leads to impaired insulin release. Therefore, identifying agents that can restore or potentiate insulin secretion from β-cells is of great significance in the development of anti-diabetic therapies. INS-1 cells are preferred for their high insulin content and reproducibility, while MIN6 cells are often used for their strong glucose responsiveness and resemblance to murine islets [22].
2.1.4 Cytotoxicity and Viability Assays
In preliminary screening of antidiabetic compounds, evaluating cytotoxicity and cell viability is critical to ensure selective bioactivity without harmful effects. The MTT assay, a widely used colorimetric method, estimates cellular viability based on mitochondrial dehydrogenase activity. Viable cells reduce the yellow tetrazolium salt [MTT] to insoluble purple formazan crystals, which are then solubilized and quantified spectrophotometrically [23]. However, the method can be influenced by compounds that alter mitochondrial function or increase mitochondrial mass, potentially skewing results [24]. In contrast, the lactate dehydrogenase [LDH] release assay quantifies cytotoxicity by measuring extracellular LDH released upon damage to the plasma membrane. LDH catalyzes the conversion of lactate to pyruvate, producing a detectable colorimetric change in the presence of tetrazolium salts. This method is non-destructive to live cells and suitable for real-time cytotoxicity monitoring, although it may be affected by background LDH in serum-containing media [25].
The Trypan Blue exclusion assay offers a simple and cost-effective method to differentiate viable from non-viable cells based on membrane integrity. Live cells exclude the dye, while damaged or dead cells absorb it and appear blue under light microscopy [26]. Despite its simplicity, the technique may lack sensitivity in detecting early apoptotic cells and requires manual cell counting, which can introduce user bias [27]. Together, these assays provide complementary insights into compound safety profiles. Employing them in combination enhances the reliability of cytotoxicity screening during early-stage evaluation of antidiabetic agents.
3.1.5 3D Cell Culture Models
Three-dimensional [3D] cell culture systems, such as spheroids and organoids, represent a significant step forward in modeling the native microenvironments of both pancreatic islets and adipose tissue for antidiabetic drug evaluation. In scaffold‑free setups, adipose‐derived cells or stromal vascular fractions self-assemble into spheroids characterized by homogeneous size, robust lipid accumulation, and secretion of adipokines like adiponectin-traits that closely parallel native adipocyte biology and improve differentiation and functional outcomes compared to monolayer cultures [28,29]. These spheroids demonstrate functional responsiveness to metabolic cues, such as lipolytic stimuli [e.g., 8‑bromo‑cAMP] and lipogenic factors [e.g., oleic acid], with quantifiable changes in lipid handling and gene expression that can be harnessed for compound screening [29]. Moreover, when incorporated into microtissue formats-including macrophage co‑cultures-3D adipose constructs can emulate insulin‑resistant phenotypes, providing a more disease-relevant platform for high-throughput drug testing [30]. Meanwhile, islet-centric 3D models-ranging from alginate‑encapsulated pancreatic islets housed in perfused microfluidic chambers to pluripotent stem cell–derived islet-like organoids-offer prolonged viability, dynamic insulin secretion, and enhanced drug sensitivity compared to traditional two-dimensional or static viability models [31,32]. Advanced "islet‑on‑a‑chip" systems further augment functionality by sustaining viability and stimulating production of critical transcription factors [e.g., PDX‑1, INS‑1, INS‑2] while enabling dynamic evaluation of antidiabetic compounds and nutritional sweeteners under physiologically relevant flow conditions [33]. Overall, 3D spheroid and organoid methodologies effectively recapitulate tissue-level architecture, biochemical signalling, and functionality relevant to metabolic regulation, thereby enhancing the fidelity and throughput of screening platforms for antidiabetic agents.
3.1.6 Microfluidic “Organ-on-a-Chip” Systems
Microfluidic organ-on-a-chip platforms represent an innovative advancement in preclinical drug screening by recreating the complex, dynamic microenvironments of key metabolic organs such as the pancreas, liver, and skeletal muscle. These miniaturized devices integrate living cells within microengineered chambers that allow precise control over fluid flow, biochemical gradients, and mechanical cues, thereby mimicking physiological conditions more accurately than traditional static cultures [34]. Specifically, pancreatic islet-on-a-chip systems enable real-time monitoring of insulin secretion dynamics in response to glucose and pharmaceutical agents, reflecting native tissue functionality with enhanced sensitivity [33]. Meanwhile, liver-on-a-chip models facilitate the assessment of drug metabolism and hepatotoxicity by sustaining hepatocyte viability and functionality under continuous perfusion [35]. Muscle-on-a-chip devices simulate insulin-stimulated glucose uptake and contractility, providing insights into peripheral insulin sensitivity and drug effects on muscle tissue [34]. The integration of multiple organ modules into interconnected microfluidic platforms further allows investigation of systemic drug responses and inter-organ crosstalk, which is critical for understanding complex metabolic disorders such as diabetes [36]. Collectively, these organ-on-a-chip technologies offer dynamic, physiologically relevant environments that improve the predictive accuracy of antidiabetic drug efficacy and toxicity screening while reducing reliance on animal models.
3.1.7 Human Induced Pluripotent Stem Cell [iPSC]-Derived β-cells
Human induced pluripotent stem cell [iPSC]-derived β-cells offer a transformative approach for modeling diabetes and screening antidiabetic drugs, as they enable the generation of patient-specific insulin-producing cells in vitro. These cells are reprogrammed from adult somatic cells and subsequently differentiated into functional β-like cells that exhibit key features of pancreatic β-cells, including glucose-stimulated insulin secretion [37]. By capturing the genetic background of individual patients, iPSC-derived β-cells provide a personalized platform to investigate variations in drug responsiveness and disease mechanisms, particularly for monogenic and complex forms of diabetes [38]. Moreover, these models allow high-throughput screening of novel compounds while reducing the ethical and practical limitations associated with primary human islets. Advances in differentiation protocols and 3D culture techniques have further enhanced the maturation and functional performance of iPSC-derived β-cells, bringing in vitro models closer to native islet physiology [39]. Consequently, iPSC technology holds promise not only for drug discovery but also for personalized medicine applications in diabetes management.
3.1.8 High-throughput Screening [HTS]
Platforms High-throughput screening [HTS] platforms play a critical role in the early-stage identification of antidiabetic compounds by allowing rapid, automated testing of large chemical libraries against disease-relevant targets or cellular models. These systems utilize miniaturized assay formats-typically in 96-, 384-, or 1536-well plates-combined with robotic liquid handling, real-time detection systems, and sophisticated data analysis software to assess multiple biological parameters in parallel [40]. In diabetes research, HTS has been applied to a variety of targets, including insulin receptor agonists, glucose transporters [e.g., GLUT4], and modulators of β-cell function or insulin secretion [41]. Modern HTS platforms are increasingly integrated with phenotypic screening using live-cell imaging, fluorescent biosensors, or reporter gene systems that capture complex, multiparametric cellular responses such as mitochondrial health, oxidative stress, and glucose uptake [42]. Additionally, coupling HTS with disease-relevant models-such as iPSC-derived β-cells or 3D spheroids-enhances biological relevance and translational potential. The evolution of high-content screening [HCS], a subtype of HTS that allows for image-based analysis of thousands of compounds, further expands the capacity to identify novel therapeutic candidates with minimal off-target effects. Overall, HTS platforms provide a scalable, efficient, and data-rich approach to discovering new agents for diabetes treatment and understanding compound mechanisms of action.
2.2 IN VIVO EXPERIMENTAL MODELS FOR ANTIDIABETIC DRUG SCREENING:
Bridging Mechanistic Insight and Translational Relevance In vivo models remain indispensable for preclinical antidiabetic drug evaluation, offering critical insights into whole-body metabolic responses, pharmacokinetics, and long-term therapeutic efficacy. These models span a wide spectrum-from chemically induced and genetic to dietary and surgically altered animals-each contributing uniquely to the understanding of diabetes pathophysiology. Chemically induced models, such as those using streptozotocin [STZ] or alloxan, selectively destroy pancreatic β-cells, resulting in hyperglycemia and mimicking type 1 diabetes-like conditions [43]. On the other hand, genetic models such as db/db mice [leptin receptor mutation], ob/ob mice [leptin deficiency], and Zucker Diabetic Fatty [ZDF] rats simulate aspects of type 2 diabetes, including obesity, insulin resistance, and β-cell dysfunction [44]. Diet-induced rodent models, particularly those fed a high-fat or high-sugar diet, closely mimic the metabolic disturbances seen in human type 2 diabetes, such as low-grade inflammation and peripheral insulin resistance [45]. Additional models include partial pancreatectomy and spontaneously diabetic strains like the Goto-Kakizaki [GK] rat, which are useful for studying β-cell adaptation in non-obese diabetic settings [46]. Modern advancements have further enriched the toolkit with CRISPR/Cas9-engineered models that allow precise replication of human diabetes mutations, enabling gene-specific intervention studies [47]. Meanwhile, zebrafish models have gained traction for their transparency, rapid development, and scalability in high-throughput screening of antidiabetic compounds [48]. Finally, humanized mouse models, generated by engrafting human islets or metabolic tissues, bridge the translational gap by providing physiologically relevant human responses to candidate therapies [49]. Collectively, these in vivo systems serve as foundational platforms for dissecting complex disease mechanisms and for evaluating the efficacy and safety of new therapeutic agents targeting diabetes.
2.2.1 Chemically Induced Models
Chemically induced animal models have long served as foundational tools for investigating diabetes pathophysiology and screening antidiabetic agents, primarily due to their ability to produce rapid and reproducible hyperglycemia. Two of the most widely used diabetogenic agents are streptozotocin [STZ] and alloxan, both of which selectively target pancreatic β-cells through oxidative stress and DNA alkylation mechanisms. STZ, a nitrosourea derivative, is taken up via the glucose transporter GLUT2, leading to DNA fragmentation, β-cell necrosis, and insulin deficiency, making it particularly effective for modeling type 1 diabetes in rodents [43]. Its dose and administration protocol can also be fine-tuned to partially damage β-cells, thereby replicating features of type 2 diabetes when combined with a high-fat diet [50]. Alloxan, another β-cell-specific toxin, acts primarily by generating reactive oxygen species [ROS] within pancreatic islets, leading to rapid β-cell apoptosis [51]. Although effective, alloxan's diabetogenic effect is more variable and species-dependent compared to STZ. These models are particularly valuable in preclinical drug screening for assessing the efficacy of insulin sensitizers, insulin secretagogues, and β-cell protective agents. However, their limitation lies in the non-progressive nature of induced diabetes and the lack of immune-mediated pathology, which reduces their utility for studying autoimmune mechanisms seen in human type 1 diabetes [43,44].
1) Diet-Induced Models
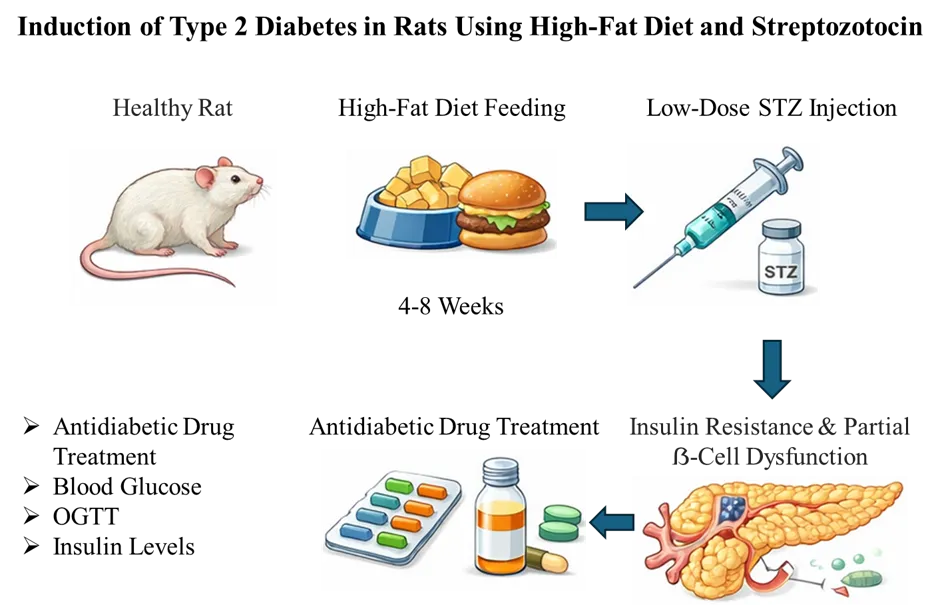
Figure 1. Schematic representation of the high-fat diet/streptozotocin (HFD–STZ)-induced Type 2 Diabetes Mellitus rat model and treatment protocol.
Diet-induced models have become highly relevant in diabetes research due to their ability to simulate the gradual onset of metabolic dysfunction seen in humans. Among these, high-fat diet [HFD]-induced models are widely used to study insulin resistance, obesity, and type 2 diabetes [T2DM] in rodents. Feeding mice or rats a diet containing 45–60% of calories from fat over several weeks leads to progressive weight gain, dyslipidemia, hepatic steatosis, and peripheral insulin resistance-hallmarks of human metabolic syndrome and early-stage T2DM [45]. These models closely mimic the environmental and lifestyle factors that contribute to the disease in humans, making them particularly valuable for evaluating insulin sensitizers, metabolic modulators, and preventive therapies. Furthermore, HFD-fed models can be combined with low-dose streptozotocin [STZ] to induce partial β-cell dysfunction, enhancing their utility for studying late-stage T2DM [52]. Their translational relevance is further improved by the use of specific diet formulations that replicate Western-style diets rich in both saturated fats and refined carbohydrates [53]. However, variability in strain susceptibility, diet composition, and feeding duration can affect outcomes, necessitating standardization for reproducibility [54]. Overall, HFD-induced models offer a robust and physiologically relevant system for screening antidiabetic agents and investigating mechanisms underlying diet-induced metabolic derangements.
2) Nicotinamide-Modified Chemically Induced Models
To develop more physiologically relevant rodent models of type 2 diabetes mellitus [T2DM], researchers have incorporated nicotinamide, a β-cell protective agent, alongside low doses of streptozotocin [STZ] or alloxan. Nicotinamide acts by inhibiting the activation of poly[ADP-ribose] polymerase [PARP], thereby mitigating oxidative DNA damage and apoptosis in pancreatic β-cells [46 55]. When administered prior to or shortly after a diabetogenic agent, it partially preserves β-cell function and insulin secretion, inducing a state of moderate hyperglycemia that mimics early-stage T2DM more closely than complete β-cell destruction models [56,57]. This approach enables the study of both insulin resistance and β-cell dysfunction, two central features of T2DM pathophysiology. The degree of β-cell impairment and glycemic imbalance can be fine-tuned by adjusting the dosing and timing of the agents, offering flexibility and reproducibility across experimental designs [58]. These models are especially valuable for screening drugs such as insulin sensitizers, GLP-1 analogs, and β-cell protectants [59]. However, limitations remain, including variability in strain sensitivity and the absence of a progressive, chronic disease course seen in human T2DM [54,55]. Nonetheless, the STZ–nicotinamide and alloxan–nicotinamide models represent widely used, cost-effective tools in preclinical antidiabetic drug evaluation.
Table 3. Common Chemical Diabetogens and Their Effective Doses Across Animal Species.
|
Chemical Agent |
Animal Species |
Typical Dose [mg/kg] |
Administration Route |
Purpose/ Remarks |
|
Streptozotocin [STZ] |
Rat |
Between 40 and 70, usually 60 |
Intraperitoneal [IP] |
Single injection to induce insulin-deficient diabetes |
|
Streptozotocin [STZ] |
Mouse |
Approximately 40, administered daily for 5 days |
Intraperitoneal [IP] |
Multiple low doses to simulate autoimmune β-cell destruction |
|
Streptozotocin [STZ] |
Rabbit |
Ranges from 25 to 50 |
Intravenous [IV] or IP |
Dose adjusted for species sensitivity in diabetes modeling |
|
Streptozotocin [STZ] |
Dog |
Starting at ~30, titrated carefully |
Intravenous [IV] |
Doses carefully adjusted to prevent toxic side effects |
|
Alloxan |
Rat |
100 to 150, typically 120 |
Intraperitoneal [IP] or IV |
Used for selective destruction of pancreatic β-cells |
|
Nicotinamide combined with STZ |
Rat |
Nicotinamide 230 prior to STZ 60 |
Intraperitoneal [IP] |
Partial β-cell impairment to mimic Type 2 diabetes |
2.2.2 Other In Vivo Models: Pancreatectomy and Spontaneously Diabetic Rodents
Beyond chemically and genetically induced models, additional in vivo systems such as pancreatectomy models and spontaneously diabetic rodent strains contribute significantly to the study of diabetes and antidiabetic drug screening. Pancreatectomy, involving partial or total surgical removal of pancreatic tissue, induces insulin deficiency by directly eliminating β-cell mass, thereby mimicking aspects of type 1 diabetes or advanced β-cell failure [60,61]. This model is instrumental in investigating β-cell regeneration, islet transplantation, and insulin replacement therapies [60,62]. However, pancreatectomy procedures are technically challenging, invasive, and prone to inter-animal variability, limiting their widespread use [61].
In contrast, spontaneously diabetic strains, notably the Goto-Kakizaki [GK] rat, provide a non-invasive, genetically stable model of type 2 diabetes characterized by impaired insulin secretion and peripheral insulin resistance without the confounding effects of obesity [63,64]. The GK rat develops chronic and progressive hyperglycemia from an early age, attributed primarily to β-cell dysfunction and reduced β-cell mass, closely reflecting human T2DM pathophysiology [65,66]. This stable diabetic phenotype facilitates long-term pharmacological studies aimed at preserving β-cell function, improving insulin sensitivity, and understanding disease progression [55]. Furthermore, GK rats exhibit diabetic complications such as nephropathy and neuropathy, providing valuable insights into secondary disease processes [62,67]. Collectively, these models expand the preclinical drug screening toolkit by offering complementary approaches to study diverse mechanisms underlying diabetes and evaluate therapeutic efficacy in physiologically relevant contexts [62,64].
1) Genetic Models
Genetic animal models provide consistent and inheritable diabetic phenotypes, making them invaluable for preclinical evaluation of antidiabetic agents. The db/db mouse, first described by Hummel et al. [1972] [68,1], harbors a mutation in the leptin receptor gene [Lepr^db] leading to leptin resistance, obesity, and progressive hyperglycemia, and remains a gold standard for modeling type 2 diabetes mellitus [T2DM] [68]. More recent studies have further characterized its metabolic and β-cell dysfunction, highlighting its suitability for testing β-cell protective therapies [69,70]. Similarly, the ob/ob mouse, with a mutation in the leptin gene [Lep^ob] causing leptin deficiency and severe obesity, continues to serve as a robust model for obesity-driven insulin resistance and glucose intolerance [71,72]. The Zucker Diabetic Fatty [ZDF] rat, carrying the fa/fa mutation in the leptin receptor gene, develops obesity and T2DM phenotypes exacerbated by a high-fat diet, providing an effective model for metabolic syndrome and diabetic complications [73,74]. Recent work has expanded understanding of its pathophysiology, including dyslipidemia and inflammation, making it highly relevant for screening drugs targeting multiple facets of metabolic disease [75]. While these genetic models offer high translational value, their monogenic origins limit full recapitulation of human T2DM’s complex etiology. Nonetheless, their well-characterized diabetic phenotypes make them essential tools in early-stage drug discovery and mechanistic research [69,75].
2) CRISPR/Cas9-Engineered Animal Models
CRISPR/Cas9 technology has enabled the development of sophisticated animal models that replicate human-specific genetic mutations underlying diabetes, thereby enhancing the translational relevance of preclinical research. For example, knockout of the insulin gene [INS] in pigs via CRISPR/Cas9 and somatic cell nuclear transfer has produced insulin-deficient piglets exhibiting hyperglycemia and glucosuria-closely emulating key diabetic phenotypes [76]. In rodent models, introduction of the human‑associated PTPN22 R620W variant into NOD mice accelerated autoimmune beta-cell destruction and heightened diabetes susceptibility, demonstrating how precise gene edits can model polygenic disease risk [77]. Similarly, CRISPR-generated Lepr knockout rats displayed obesity, hyperphagia, insulin resistance, and glucose intolerance, overcoming limitations of traditional models like Zucker rats [78]. Beyond rodents, large animal models such as pigs-sharing human-like islet structure, insulin sequence, and incretin hormone conservation-are ideal for studying type 2 diabetes physiology and pharmacology [79]. CRISPR/Cas9 also facilitated mutation of HNF1A in human embryonic stem cell [hESC]-derived beta cells to mimic MODY3, revealing impaired insulin expression, dysfunctional endocrine differentiation, and a human-specific increase in alpha-cell markers-phenotypes not observed in heterozygous knockout mice [80]. Collectively, these models underscore CRISPR/Cas9’s precision in recreating human monogenic and polygenic diabetic mutations across species, offering robust platforms for mechanistic insight, drug screening, and translational studies in antidiabetic therapy development.
3) Zebrafish Diabetes Models
Zebrafish [Danio rerio] have emerged as powerful models in diabetes research due to their optical transparency, rapid development, and amenability to genetic manipulation. These features make them ideally suited for in vivo phenotypic screening of antidiabetic compounds. For instance, transgenic zebrafish lines engineered to express fluorescent β-cell markers-combined with β-cell ablation systems utilizing nitroreductase [NTR]/ metronidazole-enable direct visualization of β-cell destruction and regeneration dynamics [81,82]. In one high-throughput screen using a pck1:Luc2 reporter line, over 2,400 bioactive compounds were tested for effects on gluconeogenic gene expression; this screen successfully identified novel glucose-lowering agents, one of which demonstrated efficacy in murine models [83]. Furthermore, chronic immersion of adult zebrafish in high-glucose solutions induces stable hyperglycemia and insulin resistance rapidly, with capacity to evaluate pharmacologic interventions such as metformin and glimepiride [84,85]. Genetic approaches using CRISPR/Cas9-targeting insulin receptor isoforms [e.g., insra, insrb] or pdx1-have generated diabetic phenotypes including elevated postprandial glucose and retinal complications, further reinforcing zebrafish utility in modeling human-like diabetes [86,87]. Together, these traits position zebrafish as powerful, high-throughput, and physiologically relevant platforms for elucidating diabetes pathways and accelerating antidiabetic drug discovery.
4) Humanized Mouse Models
Humanized mouse models-immunodeficient mice engrafted with human cells, tissues, or immune systems-have emerged as invaluable tools for diabetes research due to their enhanced physiological and immunological fidelity compared to conventional rodent models. For instance, NOD-scid IL2rγ^null [NSG] mice are profoundly immunodeficient, enabling robust engraftment of human hematopoietic stem cells [HSCs], islets, and functional human immune compartments [e.g., Hu-SRC-SCID, BLT] to investigate human-specific autoimmune and islet pathophysiology [88,52]. Specific models such as NRG-Akita [bearing the Ins2^Akita mutation] spontaneously develop hyperglycemia-and upon transplantation of human islets-can restore normoglycemia, offering a dynamic platform to assess human β-cell function and immune-mediated rejection in vivo [89]. Moreover, these mice allow detailed dissection of early disease mechanisms and pathogenesis in type 1 diabetes, including infiltration of human immune cells into the pancreas-capabilities that are ethically or technically infeasible in human patients [52,90]. While these models bridge a critical translational gap, challenges remain-such as MHC mismatches, human immune cell education on the mouse thymus, and incomplete recapitulation of cytokine signaling-that require thoughtful design and interpretation in preclinical research [91,92]. Nonetheless, humanized mouse models provide a robust, human-relevant platform for drug screening, immunological investigation, and therapeutic validation for diabetes.
5) Multi‑Organ Interaction Models
Advances in microphysiological systems-also known as organ-on-a-chip or multi-organ platforms-have provided sophisticated in vitro models that capture the dynamic metabolic interactions among key organs implicated in diabetes, including the liver, pancreas, adipose tissue, and muscle. For instance, a three-organ system integrating pancreatic β-cell, skeletal muscle, and liver modules was shown to reproduce physiological glucose–insulin dynamics by mathematically tuning interorgan connectivity and achieving realistic glucose homeostasis [93]. Similarly, a humanized model co-culturing pancreatic islet microtissues with liver spheroids demonstrated robust, prolonged endocrine crosstalk; glucose-stimulated insulin secretion from islets led to enhanced glucose uptake by liver spheroids, effectively mimicking a glucose tolerance test in vitro [94]. Another study developed a microfluidic device linking liver and adipose tissue compartments-including white and brown adipocytes-to explore mechanisms of insulin resistance. Interestingly, the presence of brown adipocytes improved hepatic insulin sensitivity and reduced de novo lipogenesis, highlighting critical adipose–liver endocrine interaction [95]. Collectively, these multi-organ models enable real-time interrogation of inter-tissue metabolic regulation, offering powerful platforms for antidiabetic drug screening and mechanistic studies in type 2 diabetes.
CONCLUSION
Advancing antidiabetic drug development hinges on the use of preclinical models that accurately mirror human metabolic physiology and disease complexity. Innovative in vitro approaches-such as organ-on-a-chip systems, human iPSC-derived β-cells, and high-throughput screening platforms-have transformed early-stage drug testing by offering dynamic, ethically sound, and patient-relevant models. At the same time, in vivo models, ranging from chemically induced and diet-based systems to genetically engineered and CRISPR-modified animals, continue to offer essential insights into whole-body responses, disease progression, and therapeutic efficacy. The integration of emerging tools-like humanized mouse models, zebrafish systems, and multi-organ microphysiological platforms-further strengthens the translational bridge between bench research and clinical application. Together, this diverse array of preclinical models forms a comprehensive and synergistic foundation for identifying, optimizing, and validating next-generation antidiabetic therapies tailored to the complexity of human diabetes.
REFERENCES

Rushikesh Choudhari, Mahesh Manke, Shubham Turewale, Padmaja Giram, Screening Approaches for Antidiabetic Agents: Integrating Classical and Advanced Methodologies, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 2812-2830. https://doi.org/10.5281/zenodo.20629593
 10.5281/zenodo.20629593
10.5281/zenodo.20629593