
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Goa College of Pharmacy, 18th June Road, Panaji, Goa, 403001, India.
The increasing prevalence of azole-resistant Candida infections necessitates the development of novel antifungal chemotypes. A series of 2,5-disubstituted-4-thiazolidinone derivatives was synthesized via a three-step protocol and structurally characterized by IR, ¹H-NMR and LC–MS. Molecular docking against Candida albicans lanosterol 14-?-demethylase (CYP51, PDB ID: 5V5Z) revealed enhanced binding affinities (?G = –37.7 to –39.3 kJ mol?¹) compared with fluconazole (–34.7 kJ mol?¹). An OECD-compliant consensus ensemble QSAR model developed from curated CYP51 inhibitor data (n = 35) demonstrated strong predictive performance (Accuracy = 91.43%, MCC = 0.7485) under leave-one-out cross-validation. Applicability domain analysis confirmed the reliability of prospective predictions. In silico ADMET profiling identified compound III a-3 as non-hepatotoxic and Ames-negative. In vitro antifungal evaluation (agar diffusion, 100 ?g disc?¹) demonstrated measurable activity against C. albicans and C. tropicalis. Collectively, the results support the potential of 2,5-disubstituted-4-thiazolidinones as CYP51-targeted antifungal candidates.
Invasive fungal infections represent a serious and escalating global health threat, accounting for more than 1.5 million deaths annually worldwide.1 Among fungal pathogens, species of the genus Candida, particularly Candida albicans and the increasingly prevalent Candida tropicalis, are major causes of opportunistic and systemic infections in immunocompromised and hospitalized patients.2,3 The azole antifungal agents remain the cornerstone of therapy and exert their effect by inhibiting lanosterol 14-α-demethylase (CYP51), a cytochrome P450–dependent enzyme essential for ergosterol biosynthesis and fungal cell membrane integrity.4 However, extensive clinical use has resulted in the emergence of resistant strains, frequently mediated by ERG11 mutations reducing azole efficacy.5 The growing incidence of resistance highlights the urgent need for structurally novel CYP51 inhibitors with improved binding characteristics and safety profiles.
Heterocyclic scaffolds are considered privileged structures in medicinal chemistry due to their tunable electronic properties and broad pharmacological relevance. Among these, 4-thiazolidinones are thiazolidine derivatives bearing a carbonyl group at the 4-position, with structural variability primarily at the 2-, 3-, and 5-positions. Substitution at these positions, particularly at C2, significantly influences the physicochemical properties and biological activity of the scaffold.6 Furthermore, 4-thiazolidinones are associated with a wide spectrum of pharmacological activities, including antimicrobial and antifungal effects, highlighting their synthetic and therapeutic relevance.7 The presence of sulfur and nitrogen atoms within the heterocycle provides favorable coordination potential toward heme-containing enzymes such as CYP51.8 Accordingly, the exploration of 2,5-disubstituted-4-thiazolidinone derivatives represents a rational and literature-supported strategy for the development of alternative antifungal chemotypes capable of addressing azole resistance.
The aim of the present research is to design and synthesize a novel series of 2,5-disubstituted-4-thiazolidinone derivatives and to evaluate their antifungal potential through a multidisciplinary strategy. The study encompasses the three-step synthesis and structural characterization of the target compounds using IR, 1H-NMR and LC–MS techniques, followed by structure-based molecular docking to investigate their interaction with the CYP51 active site. In addition, an OECD-compliant (Organisation for Economic Co-operation and Development) consensus machine learning QSAR model was developed using curated CYP51 inhibitor data to predict biological activity and assess the reliability of the proposed chemotype within its applicability domain. Drug-likeness and safety were further examined through in silico ADMET profiling, and the antifungal efficacy was experimentally evaluated against Candida albicans and Candida tropicalis.
EXPERIMENTAL
Materials and Instrumentation
All reagents and solvents were purchased from Loba Chemie, Molychem, Chemsworth, Alfa Aesar and Qualigens and used without further purification. Melting points were determined in open capillary tubes using a digital melting point apparatus EI 934 and were left uncorrected. The progress of the reactions and the purity of the synthesized compounds were monitored by thin-layer chromatography (TLC) on silica gel G-coated plates using Ethyl acetate:Chloroform (7:3) as the mobile phase and spots visualized using Meta-Lab MSI-85 UV-chamber at short wavelength. Infrared (IR) spectra were recorded on Shimadzu IR Affinity – 1S Fourier – transform infrared spectrophotometer. ¹H-NMR spectra were recorded on Bruker Advance Neo 500 NMR spectrometer using DMSO-δ6 as the solvent and TMS as the internal standard. Mass spectra (LC-MS) were obtained from Waters, SYNAPT – XS HDMS DBA064 mass spectrometer.
The synthetic strategy for the preparation of 2,5-disubstituted-4-thiazolidinone derivatives was adapted from previously reported methodologies for thiazolidinone synthesis, with modifications in the choice of aromatic aldehydes to generate structurally distinct derivatives.9
Synthesis of 2,5-Disubstituted-4-thiazolidinones.
Step 1: Synthesis of 2-chloro-N-(substituted phenyl) acetamides (Ia – Ie). Previously cooled chloroacetyl chloride (0.023 moles) was added dropwise to substituted anilines (0.01 moles) under anhydrous conditions and stirred for 4 hours at room temperature. Sodium bicarbonate solution was added to neutralize the residue and the contents were filtered off and thoroughly washed with cold water. It was collected and recrystallized with ethanol.
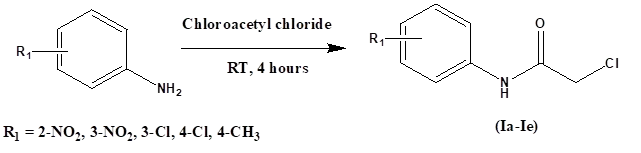
Scheme 1. Synthesis of 2-chloro-N-(substituted phenyl) acetamides (Ia – Ie).
Step 2: Synthesis of 2-(substituted phenylimino)-4-thiazolidinones (IIa – IIb). In a round bottom flask previously synthesized 2-chloro-N-(substituted phenyl) acetamides (Ia – Ie) (0.005 moles) was refluxed with ammonium thiocyanate (0.01 moles), using ethanol as solvent, for 4 – 5 hours. The reaction mixture was allowed to stand overnight. The precipitate was filtered off, washed with water and recrystallized from ethanol.

Scheme 2. Synthesis of 2-(substituted phenylimino)-4-thiazolidinones (IIa – IIb).
Step 3: Synthesis of 2-(substituted phenylimino)-5-(substituted arylidene)-4-thiazolidinones (III a-1 – III a-3). In a clean, dry round bottom flask, previously synthesized 2-(substituted phenylimino)-4-thiazolidinones (IIa – IIb) (0.004 moles), glacial acetic acid (0.4 moles) and sodium acetate (0.008 moles) was stirred. Aromatic aldehydes (0.006 moles) – 3,4-dimethoxybenzaldehyde, 2,4-dimethoxybenzaldehyde and 2,4-dihydroxybenzaldehyde – were added and the mixture was refluxed for 8 – 10 hours. The precipitate was obtained after cooling the reaction mixture to room temperature and was subsequently filtered, washed thoroughly with water and recrystallized using methanol as a solvent.
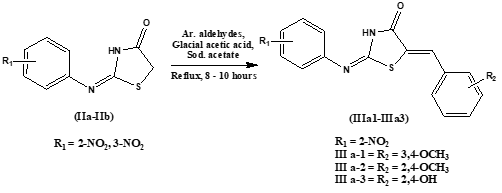
Scheme 3. Synthesis of 2-(substituted phenylimino)-5-(substituted arylidene)-4-thiazolidinones (III a-1 – III a-3).
Computational Methodology
Protein Preparation.
The high-resolution crystal structure of Candida albicans lanosterol 14 alpha-demethylase (CYP51) complexed with a tetrazole inhibitor was obtained from the RCSB Protein Data Bank (PDB ID: 5V5Z).10 Initial processing was conducted using BIOVIA Discovery Studio Visualizer v26.1.0.24284, where the protein structure was cleaned by removing co-crystallized water molecules, heteroatoms, and the native ligand. The Heme-601 prosthetic group and its central iron (Fe) atom were specifically retained as essential components of the catalytic site.
The simplified protein-heme complex was then imported into AutoDock Tools (ADT) v.1.5.7.11 Within ADT, polar hydrogen atoms were added to the structure, and Kollman charges were assigned to the macromolecule. The iron center within the heme was preserved to facilitate metal-coordination studies. The finalized receptor was saved in PDBQT format (Protein Data Bank, Partial Charge & Atom Type), defining the essential coordinates and partial charges required for the molecular docking simulations.
Molecular Docking Validation.
To ensure the reliability of the docking protocol and the accuracy of the charged heme receptor model, a self-docking validation was performed. The co-crystallized tetrazole inhibitor was extracted from the 5V5Z complex to serve as the reference ligand. Ligand preparation was conducted by adding hydrogen atoms and performing geometry optimization using the MMFF94 force field12 via the RDKit library. Subsequently, OpenBabel13 was employed to assign Gasteiger partial charges and adjust the protonation state to physiological pH (7.4).
The active site was defined by calculating the heavy-atom centroid of the native ligand coordinates. Molecular docking was executed using AutoDock Vina v.1.1.214 with a grid box centered at x = -37.516, y = -17.458, and z = 26.227. To ensure an exhaustive search of the conformational space, the box dimensions were set to 20 × 20 × 20 Å, and the exhaustiveness parameter was increased to 32.
The validation yielded a top-ranked docking pose with a binding affinity of –51.9 kJ mol⁻¹. The Root Mean Square Deviation (RMSD) between the redocked pose and the original co-crystallized orientation was calculated to be 1.930 Å. Since the RMSD was below the widely accepted threshold of 2.0 Å, the docking parameters and the receptor model were considered validated and this established protocol was subsequently applied for the docking of the synthesized 2,5-disubstituted-4-thiazolidinone derivatives.
Ligand Preparation.
The 2D structures of the synthesized 2,5-disubstituted-4-thiazolidinone derivatives (III a-1 – III a-3) and the reference drug, fluconazole, were drafted using ChemDraw Pro 8.0. The structures were converted into Simplified Molecular Input Line Entry System (SMILES) strings for computational processing.
Three-dimensional (3D) coordinates were generated from the SMILES strings using OpenBabel v.3.1.1. A systematic 3D structure generation was performed with the best parameter to ensure the selection of the lowest-energy conformer. To simulate physiological conditions, the protonation states of the ligands were adjusted to pH 7.4.
Partial charges were assigned using the Gasteiger-Marsili method.15 The finalized ligands were parameterized by merging non-polar hydrogen atoms and defining rotatable bonds. The processed structures were saved in PDBQT format for subsequent molecular docking simulations against the CYP51 target.
Molecular Docking.
Molecular docking simulations were performed to evaluate the binding affinities and interaction modes of the synthesized 2,5-disubstituted-4-thiazolidinone derivatives (III a-1 – III a-3) and the reference standard, fluconazole, against the C. albicans CYP51 target. The simulations were carried out using AutoDock Vina v.1.1.2. The screening utilized the previously validated grid parameters and search space.
The resulting docking poses were ranked based on their predicted binding free energies (ΔG, kcal mol-1). The top-ranked pose for each derivative, characterized by the lowest binding energy, was selected for further interaction analysis. Visual assessment of the protein-ligand complexes was performed using the Protein-Ligand Interaction Profiler (PLIP)16 and Pymol to identify critical hydrogen bonding, and π-stacking.
Data Acquisition and Curation for QSAR.
A focused dataset of Candida albicans lanosterol 14-α-demethylase (CYP51) inhibitors was retrieved from the BindingDB database.17 Reported IC₅₀ values were converted to pIC₅₀ (log₁₀IC₅₀, M) for uniformity. Compounds were classified into binary categories based on activity thresholds: molecules with significant inhibitory activity were labeled as active (1), while compounds lacking meaningful inhibition were labeled as inactive (0).
Chemical curation included removal of duplicates, salts, and inconsistent entries, followed by standardization of SMILES representations. Molecules with invalid or unparsable SMILES were excluded. After curation, the final dataset consisted of 35 structurally diverse compounds (27 actives and 8 inactives), ensuring chemical consistency for model development.
Molecular Descriptor Generation.
Structural features were encoded using 166-bit MACCS (Molecular Access System) fingerprints generated via RDKit. MACCS keys were selected due to their interpretability and ability to capture predefined functional fragments relevant to CYP51 inhibition. The binary fingerprint matrix served as the descriptor space for subsequent machine learning modelling.
Consensus Ensemble Model Development.
To minimize algorithmic bias and improve predictive robustness, a consensus ensemble strategy was employed.18, 19 Three complementary machine learning classifiers were integrated:
Hyperparameter optimization was performed through grid exploration of tree depth, Support Vector Machine (SVM) regularization parameter (C), and voting strategy (soft vs hard voting). Model selection was based on maximizing the Matthews Correlation Coefficient (MCC), which provides a balanced performance metric for imbalanced binary datasets.
The final consensus model employed soft voting with optimized parameters (tree depth = 3; SVM C = 0.01), combining probabilistic outputs of the individual classifiers.
Model Validation.
Given the limited dataset size, Leave-One-Out Cross-Validation (LOOCV) was applied to evaluate OECD compliance.23 In this approach, the model was iteratively trained on N−1 compounds and tested on the remaining compound until all molecules were evaluated. To assess the model's ability to correctly identify both active and inactive compounds, Sensitivity and Specificity were computed. Applicability domain (AD) assessment was performed using Tanimoto similarity based on MACCS fingerprints. Compounds with similarity values within the chemical space of the training set were considered reliable predictions.
Pharmacokinetic and Toxicity Prediction.
To evaluate the drug-likeness and safety profiles of the synthesized 2,5-disubstituted-4-thiazolidinone derivatives and the reference standard fluconazole, in silico ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiling was conducted. The Simplified Molecular Input Line Entry System (SMILES) strings for each compound were submitted to the pkCSM web server.24
Antifungal Evaluation
The synthesized 2,5-disubstituted-4-thiazolidinone derivatives (III a-1 – III a-3) were evaluated for in vitro antifungal activity against Candida albicans and Candida tropicalis using the agar well diffusion method, with fluconazole as the reference standard. Stock solutions were prepared by dissolving 10 mg of each test compound in 1 mL of dimethyl sulfoxide (DMSO). The fungal strains were cultured on Sabouraud agar medium. Inocula were prepared by transferring colonies into broth and adjusting the turbidity to match the 0.5 McFarland standard (1 × 10⁶ CFU mL⁻¹).25 The agar surface was uniformly streaked using a sterile cotton swab in three directions to ensure confluent growth, and the plates were allowed to equilibrate for three minutes before well formation.
Wells of 5 mm diameter were created in the inoculated agar using a heated hollow tube. Each well was loaded with 50 μL of the respective stock solution (test compounds and standard). The plates were incubated at 37 °C for 18–24 hours. The resulting zones of inhibition were measured in millimetres (mm) from the edge of the well to the margin of fungal growth. All assays were performed in duplicate to ensure reproducibility, and the inhibitory effects were compared against the fluconazole standard to determine the comparative potency of the synthesized derivatives.
RESULTS AND DISCUSSION
Chemistry
2-(2'-nitrophenylimino)-5-(3,4-dimethoxybenzylidene)-4-thiazolidinone (III a-1) Brown solid; Yield 60.2 %; mp 178–181 ºC; Rf = 0.84 (Ethyl acetate:Chloroform, 9:1); Solubility: DMSO; IR / cm⁻¹: 3124.68 (N–H stretching), 3007.02 (C–H stretching aromatic), 2835.36 (C–H stretching aliphatic), 1701.22 (C=O stretching lactam), 1651.07 (C=C stretching), 1589.34 (C=N stretching), 1573.91 (N–H bending), 1508.33 (N=O bending), 1436.97 (C–H bending aliphatic), 1243.73, 1018.41 (C–O–C stretching ether), 700.16 (C–S stretching); ¹H NMR (400 MHz, DMSO, δ): 3.75–3.78 (s, 6H, OCH3), 7.03–7.08 (m, 2H, Ar–H), 7.10 (s, 1H, C=C–H), 7.31–7.40 (m, 2H, Ar–H), 7.66–7.73 (m, 2H, Ar–H), 8.02–8.03 (d, 1H, Ar–H), 12.57 (s, 1H, 4-thiazolidinone NH); MS (ESI) m/z: 386.08 [M + H]⁺.
2-(2-nitrophenylimino)-5-(2,4-dimethoxybenzylidene)-4-thiazolidinone (III a-2) Orange solid; Yield 70.89 %; mp 205–207 ºC; Rf = 0.83 (Ethyl acetate:Chloroform, 9:1); Solubility: DMSO; IR / cm⁻¹: 3110 (N–H stretching), 3000 (aromatic C–H stretching), 2800 (aliphatic C–H stretching), 1700 (C=O stretching lactam), 1650 (C=C stretching), 1640 (C=N stretching), 1575 (N–H bending), 1525 (N–O bending), 1450 (aliphatic C–H bending), 1250, 1020 (C–O–C stretching ether), 700 (C–S stretching); ¹H NMR (400 MHz, DMSO, δ): 3.79–3.86 (s, 6H, OCH3), 6.62–6.64 (d, 2H, Ar–H), 7.21–7.22 (d, 1H, C=C–H), 7.28–7.29 (d, 1H, Ar–H), 7.36–7.39 (t, 1H, Ar–H), 7.70–7.72 (t, 1H, Ar–H), 7.86 (s, 1H, Ar–H), 8.02–8.03 (d, 1H, Ar–H), 12.57 (s, 1H, 4-thiazolidinone NH); MS (ESI) m/z: 386.05 [M + H]⁺.
2-(2-nitrophenylimino)-5-(2,4-dihydroxybenzylidene)-4-thiazolidinone (III a-3) Red solid; Yield 62.5 %; mp 209–211 ºC; Rf = 0.81 (Ethyl acetate:Chloroform, 9:1); Solubility: DMSO; IR / cm⁻¹: 3400–3000 (O–H stretching, N–H stretching, aromatic C–H stretching), 1650 (C=O stretching lactam), 1630 (C=C stretching), 1600 (C=N stretching), 1520 (N–H bending), 1450 (N–O bending), 700 (C–S stretching); ¹H NMR (400 MHz, DMSO, δ): 6.30 (d, 1H, Ar–H), 6.45 (d, 1H, Ar–H), 7.40 (d, 1H, Ar–H), 7.85 (s, 1H, C=C–H), 9.80 (s, 1H, OH), 10.20 (s, 1H, OH), 12.57 (s, 1H, 4-thiazolidinone NH); MS (ESI) m/z: 358.02 [M + H]⁺.
Molecular Docking and Interaction Analysis
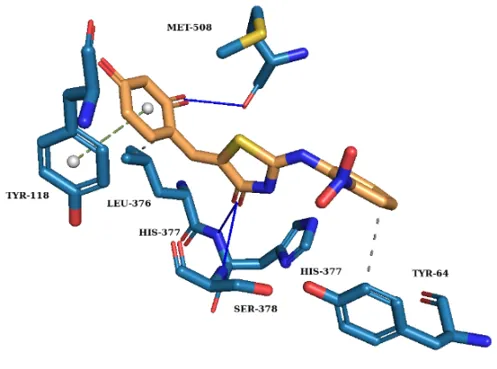
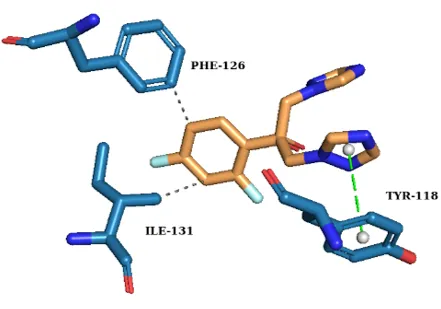
To elucidate the binding mechanism against Candida albicans CYP51, molecular docking was performed using a validated protocol into the 5V5Z active site. All synthesized derivatives exhibited higher binding affinities than fluconazole (Table I) retaining key favourable interactions (Fig. 1).
Table I. Binding affinities and molecular interactions of synthesized compounds and fluconazole.
|
Compound |
Affinity (kJ mol⁻¹) |
Hydrogen Bonds |
Hydrophobic Interactions |
π-Stacking |
|
III a-1 |
–37.7 |
His377, Ser378 |
Phe126, Phe233 |
- |
|
III a-2 |
–38.1 |
Met508 |
Phe126, Phe233, Phe380 |
- |
|
III a-3 |
–39.3 |
His377, Ser378, Met508 |
Tyr64, Leu376 |
Tyr118 |
|
FLZ |
–34.7 |
- |
Phe126, Ile131 |
Tyr118 |
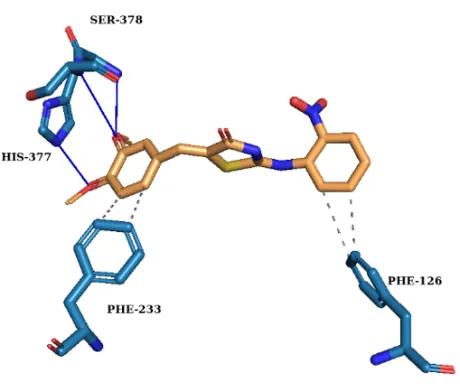
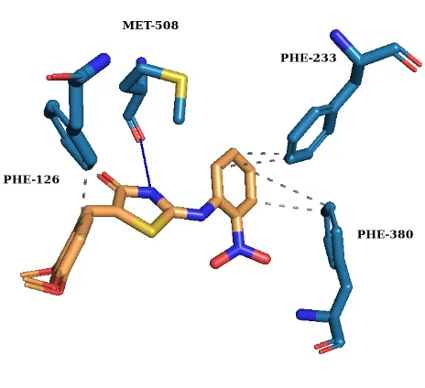
(a) (b)
(c) (d)
Fig. 1. 3D Molecular docking interactions of (a) Compound III a-1, (b) Compound III a-2, (c) Compound III a-3 and (d) Fluconazole within the active site of CYP51.
QSAR Model Performance and Predictive Reliability
The consensus ensemble model demonstrated strong predictive performance under LOOCV validation (Table II). The MCC value of 0.7485 indicates robust classification beyond random assignment and confirms balanced performance despite moderate class imbalance. The confusion matrix (Fig. 2) shows that only one active compound was misclassified (FN = 1), while two inactive compounds were incorrectly predicted as active (FP = 2). The high sensitivity (96.30%) reflects the model’s strong ability to correctly identify potent CYP51 inhibitors.
Table II. ML-QSAR performance matrices.
|
Metric |
Value |
|
Accuracy |
91.43% |
|
MCC |
0.7485 |
|
Sensitivity |
96.30% |
|
Specificity |
75.00% |
|
Precision |
92.86% |
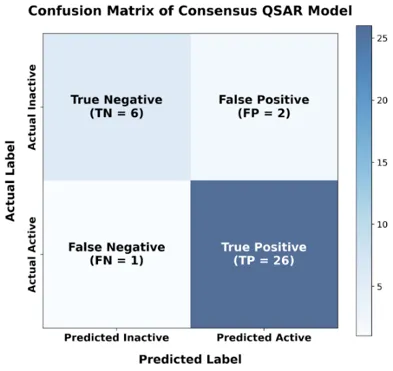
Fig. 2. Confusion matrix of consensus QSAR Model.
Prospective Prediction and Interpretation
The validated model predicted all synthesized derivatives (III a-1 – III a-3) as Active with high confidence scores (87.8–90.2%). The maximum Tanimoto similarity values (0.48–0.53) confirm that the compounds reside within the structural domain of the training dataset, supporting the reliability of the predictions. Fluconazole, used as reference, showed high similarity (1.0) to the training space, validating chemical space consistency (Table III).
Table III. QSAR Predicted Activity and Applicability Domain Assessment.
|
Compound |
Predicted Class |
Confidence (%) |
Similarity (AD) |
|
III a-1 |
Active |
90.20% |
0.53 |
|
III a-2 |
Active |
89.80% |
0.53 |
|
III a-3 |
Active |
87.80% |
0.48 |
|
FLZ |
Active |
71.40% |
1 |
Feature importance analysis derived from the Random Forest component identified MACCS_42 as the most influential descriptor (importance score = 0.1025), followed by MACCS_57 and MACCS_106. These fragments correspond to functional motifs commonly associated with heterocyclic scaffolds and aromatic substitutions, supporting the relevance of the 2,5-disubstituted-4-thiazolidinone framework in CYP51 inhibition.
ADMET Profiling and Toxicity
The drug-likeness and safety profiles were assessed to ensure the therapeutic viability of the compounds. All derivatives followed Lipinski’s Rule of Five. Notably, III a-1 and III a-3 were predicted to be non-hepatotoxic and Ames-negative. This represents a significant advantage over Fluconazole and III a-2, which both showed predicted hepatotoxicity (Table IV and Table V).
Table IV. Predicted pharmacokinetic parameters.
|
Compound |
HIA (%) |
BBB (logBB) |
CNS (logPS) |
CYP3A4 Substrate |
P-gp Substrate |
|
III a-1 |
81.02 |
-1.034 |
-2.332 |
Yes |
Yes |
|
III a-2 |
90.26 |
-0.516 |
-2.234 |
Yes |
Yes |
|
III a-3 |
90.19 |
-0.545 |
-2.224 |
No |
Yes |
|
FLZ |
94.96 |
-1.067 |
-3.185 |
No |
No |
Table V. Predicted toxicity parameters.
|
Compound |
Ames Toxicity |
Hepatotoxicity |
Skin Sensitization |
hERG I Inhibitor |
hERG II Inhibitor |
|
III a-1 |
No |
No |
No |
No |
No |
|
III a-2 |
Yes |
Yes |
No |
No |
No |
|
III a-3 |
No |
No |
No |
No |
Yes |
|
FLZ |
No |
Yes |
No |
No |
No |
Antifungal Activity
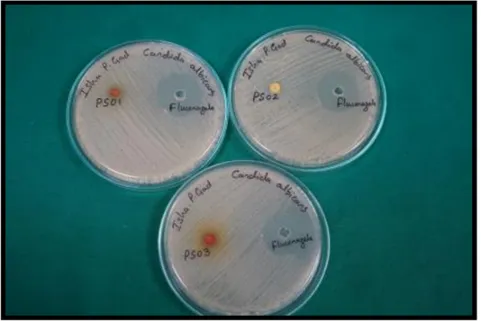
The in vitro activity results (Table VI) demonstrated that all compounds possessed antifungal activity under diffusion conditions. Although fluconazole exhibited larger zones of inhibition (ZOI), likely due to its superior cell wall penetration, the synthesized derivatives showed consistent activity against both C. albicans and C. Tropicalis (Fig. 3).
Table VI. Antifungal activity (Zone of Inhibition in mm) at 100 μg disc⁻¹.
|
Compound |
Candida albicans (ZOI in mm) |
Candida tropicalis (ZOI in mm) |
|
III a-1 |
12 |
18 |
|
III a-2 |
- |
20 |
|
III a-3 |
11 |
18 |
|
FLZ |
30 – 34 |
30 – 34 |
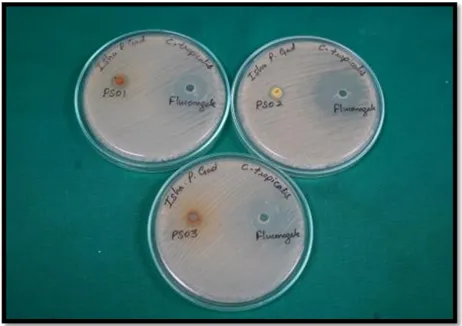
(a) (b)
Fig. 3. Antifungal activity (Zone of Inhibition in mm) at 100 μg disc⁻¹ against (a) Candida albicans and (b) Candida tropicalis.
The comparison between predicted binding affinities (ΔG, kJ mol⁻¹) and experimentally measured zones of inhibition indicates a qualitative relationship between CYP51 binding strength and antifungal response. Compound III a-3, which exhibited the lowest docking energy (–39.3 kJ mol⁻¹), also demonstrated consistent activity against Candida albicans and Candida tropicalis, supporting the relevance of the docking predictions.
Although fluconazole produced larger inhibition zones, likely due to superior diffusion and permeability characteristics, the synthesized derivatives showed stronger predicted enzyme-binding affinities. These findings suggest that docking-derived binding energies correlate with intrinsic target interaction but do not directly translate to agar diffusion outcomes, which are influenced by additional physicochemical factors. Overall, the results support the predictive value of structure-based docking while underscoring the multifactorial nature of antifungal bioactivity.
CONCLUSION
The present study describes the successful synthesis and spectroscopic characterization of a novel series of 2,5-disubstituted-4-thiazolidinone derivatives and their multidisciplinary evaluation as potential antifungal agents. Structure-based molecular docking using a validated CYP51 model demonstrated favourable binding affinities of the synthesized derivatives relative to fluconazole, supporting their potential to interact effectively within the enzyme active site.
An OECD-compliant consensus ensemble QSAR model exhibited strong predictive performance under leave-one-out cross-validation, confirming the reliability of the computational framework. Applicability domain analysis indicated that the synthesized compounds fall within the defined chemical space of the training dataset, reinforcing the credibility of the prospective predictions. In silico ADMET profiling further highlighted compound III a-3 as possessing a comparatively favourable safety profile.
In vitro antifungal screening demonstrated measurable activity against Candida albicans and Candida tropicalis, and correlation analysis revealed a qualitative relationship between docking-derived binding affinities and biological response. While diffusion-based assays are influenced by physicochemical factors beyond intrinsic enzyme binding, the combined computational and experimental findings collectively support the 2,5-disubstituted-4-thiazolidinone scaffold as a promising platform for further structural optimization toward improved antifungal efficacy.
DATA AVAILABILITY
All curated datasets, machine learning model scripts, descriptor files, model training and validation outputs, and prospective prediction results generated in this study are openly available in the GitHub repository: https://github.com/HardikKNaik/CYP51-Antifungal-QSAR-Model. The repository includes source data for the consensus QSAR model, and applicability domain analysis. Additional experimental details, high-resolution spectra, raw computational files, and other auxiliary data not included in the main manuscript are available from the corresponding author upon reasonable request.
REFERENCES

Hardik Naik, Isha Gad, Teja Walke, Vithal Bhandare, Antioxidant Activity of Hydro-Alcoholic Root Extract of Clitoria ternatea Linn. Plant, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 468-479. https://doi.org/10.5281/zenodo.21141504
 10.5281/zenodo.21141504
10.5281/zenodo.21141504