
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,4Department of Chemistry, Anurag University, Hyderabad, India.
2,3Analytical Research and Development, Aurore Life Sciences Private Limited, Hyderabad, India.
High-Performance Liquid Chromatography (HPLC) is a crucial analytical technique widely employed in the pharmaceutical industry for the identification, quantification, and control of genotoxic impurities (GTIs). These impurities, such as nitrosamines, pose significant public health risks due to their carcinogenic potential, necessitating precise detection and stringent regulatory control. This review explores the fundamentals of HPLC, including its principles, instrumentation, operational methodologies, and various applications in pharmaceutical analysis. It emphasizes the development and validation of methods tailored for detecting trace-level impurities, with a particular focus on adherence to International Council for Harmonisation (ICH) guidelines. Furthermore, the review examines advancements in HPLC technology and innovative detectors that improve analytical sensitivity and operational efficiency. By covering these critical aspects, the discussion underscores HPLC’s indispensable role in ensuring pharmaceutical quality, safeguarding patient safety, and achieving regulatory compliance
Pharmaceutical products are required to comply with stringent safety and quality standards to ensure the protection of patient health [1,2]. Among the pivotal challenges in pharmaceutical development, the identification, quantification, and control of genotoxic impurities (GTIs) represent a critical focus area [4]. GTIs, even at trace concentrations, have the potential to interact with genetic material, leading to mutations and posing significant long-term health risks, including carcinogenesis (ICH, 2017). Consequently, global regulatory authorities such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and the European Medicines Agency (EMA) have promulgated comprehensive guidelines to mitigate risks associated with GTIs. High-Performance Liquid Chromatography (HPLC) has emerged as a cornerstone analytical methodology in this context, offering unparalleled sensitivity, specificity, and robustness in the analysis of non-volatile, thermally labile, and structurally diverse impurities [3,5]. Its application extends across the detection, separation, and quantification of GTIs, enabling adherence to rigorous regulatory standards. This review elucidates the critical role of HPLC in pharmaceutical quality assurance, with a specific focus on advanced strategies for GTI detection, method optimization, analytical validation, and compliance with evolving regulatory frameworks.
High-Performance Liquid Chromatography
Principle
High-Performance Liquid Chromatography (HPLC) operates on the principle of differential partitioning of analytes between a stationary phase and a liquid mobile phase under high-pressure conditions [6,7]. The sample is introduced into the mobile phase stream and transported through a column packed with chromatographic media, typically composed of silica or polymer-based particles functionalized to provide specific chemical interactions [9]. The separation of components arises from the distinct affinities of analytes for the stationary phase, governed by physicochemical properties such as polarity, molecular weight, hydrophobicity, and ionizability [8]. As analytes traverse the column, their retention times are modulated by the strength and nature of these interactions, resulting in the temporal resolution of individual components [10]. The eluted species are detected and quantified using highly sensitive detectors, such as ultraviolet-visible (UV-Vis) absorbance, fluorescence, refractive index, or mass spectrometry, each providing precise qualitative and quantitative characterization. The versatility, high resolution, and sensitivity of HPLC render it indispensable across domains, including pharmaceutical quality control, food safety evaluation, and environmental contaminant monitoring, particularly for complex sample matrices requiring robust analytical performance.
Instrumentation:
HPLC instrumentation consists of several key components:
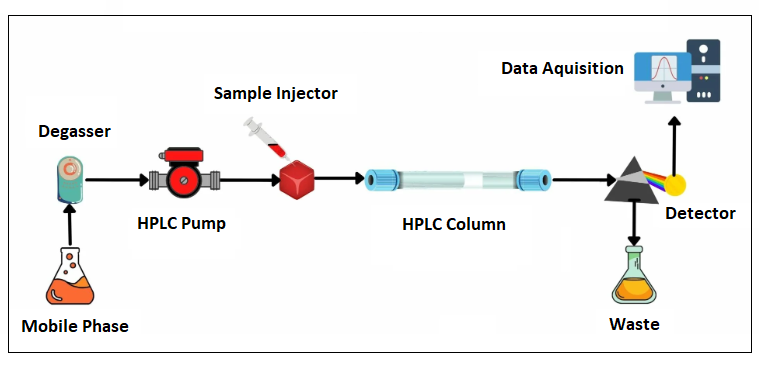
Solvent Delivery System:
High-Performance Liquid Chromatography (HPLC) utilizes a precision-engineered high-pressure pumping system to propel the mobile phase through the chromatographic column at meticulously regulated flow rates [6]. The solvent delivery mechanism supports both isocratic and gradient elution modes, chosen based on the specific analytical objectives [9]. Isocratic elution maintains a constant mobile phase composition, while gradient elution systematically varies the solvent polarity or composition over time, optimizing the separation of analytes in complex multi-component matrices [6]. Gradient elution is particularly advantageous for resolving mixtures with wide-ranging physicochemical properties, enhancing chromatographic efficiency and peak resolution.
Injection System:
The introduction of analyte samples into the mobile phase is facilitated via precision injection mechanisms, employing either automated autosamplers or manual injection loops [11]. Automated autosamplers are particularly advantageous, offering enhanced reproducibility, precision, and operational efficiency, which are critical for high-throughput analytical workflows. These systems minimize variability associated with manual sample handling and are integral to ensuring consistency in retention times and peak area measurements across multiple injections, thereby supporting the robustness of high-volume chromatographic analyses.
Columns:
The chromatographic column, packed with silica-based stationary phases functionalized with chemically specific moieties, is a pivotal component in achieving high separation efficiency [9]. Reversed-phase columns, notably octadecylsilane (C18) phases, are extensively employed in pharmaceutical analysis owing to their broad applicability and robustness. The hydrophobic nature of C18 stationary phases facilitates the separation of analytes based on polarity, making them particularly suitable for resolving a diverse array of compounds in complex matrices. Their versatility, coupled with exceptional stability under varied mobile phase conditions, underscores their prominence in modern chromatographic methodologies.
Detectors:
High-Performance Liquid Chromatography (HPLC) integrates a variety of advanced detection systems to facilitate qualitative and quantitative analysis, tailored to the physicochemical properties of target analytes.
Key detection modalities include:
• UV-Vis Detectors:
These detectors exploit the ability of chromophoric compounds to absorb ultraviolet or visible light at specific wavelengths [10]. By measuring the absorbance intensity, UV-Vis detectors provide highly reliable quantitative data, making them ideal for compounds with well-defined absorption spectra. Diode-array detectors (DAD) enhance analytical capabilities by enabling simultaneous multi-wavelength detection and spectral analysis.
• Fluorescence Detectors:
Leveraging the fluorescence properties of specific analytes, these detectors offer unparalleled sensitivity and selectivity. Fluorescence detectors are particularly effective for trace-level quantification in complex matrices, as they measure the emitted light intensity upon excitation of the analyte with a specific wavelength. Applications include the detection of compounds inherently fluorescent or derivatized to exhibit fluorescence.
• Mass Spectrometry (LC-MS):
Coupling HPLC with mass spectrometry enables comprehensive structural elucidation and precise quantification of analytes. LC-MS systems provide molecular mass determination, structural fragmentation patterns, and isotopic distribution analysis. This capability is particularly critical for characterizing complex mixtures, impurities, metabolites, and genotoxic compounds with high specificity and sensitivity. The integration of tandem mass spectrometry (LC-MS/MS) further enhances detection limits and resolution, supporting robust analysis in pharmaceutical, biological, and environmental applications.
Data Acquisition System:
High-Performance Liquid Chromatography (HPLC) incorporates advanced data acquisition systems equipped with specialized software to capture, process, and interpret chromatographic data with exceptional precision [12]. These systems continuously monitor the signal output from detectors, such as UV-Vis, fluorescence, or mass spectrometry, converting analog signals into high-resolution digital chromatograms. The software algorithms facilitate baseline correction, noise reduction, and peak integration, enabling accurate quantification of analytes and identification based on retention times and detector-specific responses. Additionally, these systems support complex analytical tasks, including multi-wavelength analysis, spectral deconvolution, and overlay comparisons, which are essential for characterizing co-eluting compounds or trace-level impurities. Modern data acquisition systems are also equipped with robust validation and compliance features, ensuring adherence to regulatory standards such as Good Laboratory Practice (GLP) and 21 CFR Part 11 for electronic data integrity. Integrated capabilities for automation, batch processing, and advanced reporting further enhance throughput and reproducibility, solidifying the data acquisition system's critical role in HPLC-based analytical workflows.
Procedure (Stepwise):
Samples are extracted and purified to remove matrix interferences. Derivatization may be required for non-chromophoric compounds. Proper sample preparation is crucial to ensure accurate and reproducible results, especially when dealing with complex matrices [13].
Mobile phase composition, flow rate, and column temperature are optimized. Optimization of these parameters is essential to achieve the desired separation efficiency and reproducibility [6]. The correct choice of mobile phase and temperature is also critical for overcoming sample matrix effects.
The sample is introduced into the system via an injector. Automated or manual injection techniques can be used, with automated systems offering better reproducibility and precision [14]. Injection volumes and accuracy are key factors for ensuring consistent chromatographic results.
Components are separated based on their interactions with the stationary phase. These interactions are influenced by various physicochemical properties such as polarity, molecular size, and charge, which determine the retention times of analytes [7].
Analytes are detected as they elute from the column. The detection method (e.g., UV-Vis, fluorescence, or mass spectrometry) is selected based on the analyte’s chemical properties, allowing for specific and sensitive detection of the compounds of interest.
Chromatograms are analyzed to quantify and identify components. Peak area, retention time, and other parameters are used for the quantitative analysis of each analyte, with software tools facilitating the integration and validation of results [12].
Use of HPLC in the Pharmaceutical Industry:
High-Performance Liquid Chromatography (HPLC) is a cornerstone analytical technique in the pharmaceutical industry, providing unmatched versatility, precision, and sensitivity for a wide array of applications in drug development, quality control, and regulatory compliance [15]. Its capability to separate, identify, and quantify components in complex mixtures makes it indispensable for ensuring the safety, efficacy, and quality of pharmaceutical products.
HPLC is extensively employed to analyze APIs in raw materials, intermediates, and finished products. The technique offers high specificity and resolution to distinguish closely related compounds, such as isomers or degradation products [15].
HPLC is pivotal in identifying and quantifying impurities, which are critical for product safety and regulatory approval. These include process-related impurities, such as residual solvents, reagents, and byproducts, as well as degradation products formed due to environmental stress or storage conditions [16]. The detection of genotoxic impurities (GTIs) at trace levels using sensitive HPLC detectors like UV-Vis or tandem mass spectrometry (LC-MS/MS) ensures compliance with stringent regulatory standards.
HPLC supports the development and validation of analytical assays critical for regulatory submissions, ensuring the reliability and reproducibility of methods [17].
HPLC plays a central role in stability studies, monitoring the chemical integrity of APIs and formulations over time under specified conditions [18]. The technique identifies and quantifies degradation products, supporting determination of shelf life and the development of storage conditions.
HPLC using chiral stationary phases (CSPs) is essential for the separation and quantification of enantiomers in chiral APIs, ensuring the safety and efficacy of drugs with chirally active components [7].
Advantages of HPLC in the Pharmaceutical Industry:
HPLC is suitable for a wide range of analytes, including polar, non-polar, and thermally labile compounds. This flexibility makes it indispensable for pharmaceutical applications involving diverse chemical structures [6].
HPLC is capable of detecting trace-level impurities and active pharmaceutical ingredients (APIs), providing reliable quantification even in complex matrices. This sensitivity is particularly critical for regulatory compliance [19].
HPLC facilitates adherence to stringent guidelines, such as those outlined by the International Council for Harmonisation (ICH), the U.S. Food and Drug Administration (FDA), and the European Medicines Agency (EMA) [20]. Its robustness ensures that the data generated meets the requirements of regulatory bodies.
HPLC is easily transferable between research and manufacturing scales, ensuring consistency across different stages of pharmaceutical development. This scalability enhances its utility in both laboratory and industrial settings [21].
HPLC is compatible with advanced detection systems like mass spectrometry (MS), diode-array detection (DAD), and fluorescence detection, significantly enhancing its analytical capabilities. This integration extends the scope of HPLC applications across various domains [7].
Types and Sources of Impurities in Pharmaceuticals
Pharmaceutical impurities arise from diverse sources during the development, manufacturing, and storage of drug products. These impurities are broadly classified into:
Process-related impurities originate during the synthesis of Active Pharmaceutical Ingredients (APIs) and include byproducts, unreacted starting materials, intermediates, and reagents. These impurities are typically the result of incomplete chemical reactions, side reactions, or impurities in the raw materials used during synthesis [22]. Analytical techniques like Gas Chromatography (GC) and High-Performance Liquid Chromatography (HPLC) are employed to detect and quantify these impurities, ensuring compliance with regulatory limits [5].
Degradation products are formed when APIs or formulations undergo chemical changes due to environmental factors such as light, heat, moisture, or oxidative conditions. These products can adversely affect the safety, efficacy, and shelf life of the drug [23]. Stability studies using GC or HPLC allow for the identification and quantification of degradation products, supporting the development of robust formulations and proper storage guidelines [22].
Contaminants are extraneous substances unintentionally introduced during manufacturing, storage, or handling. These may include microorganisms, particulate matter, or residues from packaging materials [24]. Their presence can compromise product quality and patient safety. Rigorous quality control protocols and analytical tools like GC are essential for detecting and minimizing contaminants, ensuring adherence to Good Manufacturing Practices (GMP) [25].
Classification Based on ICH Guidelines
The International Council for Harmonisation (ICH) has established comprehensive guidelines for classifying pharmaceutical impurities into three primary categories: organic impurities, inorganic impurities, and residual solvents. These classifications are crucial in the pharmaceutical industry, particularly when applying analytical techniques such as High-Performance Liquid Chromatography (HPLC), which is instrumental in identifying and quantifying these impurities to ensure the safety and quality of pharmaceutical products [26].
Organic impurities are classified into process-related impurities and degradation products. These impurities can emerge during the synthesis of Active Pharmaceutical Ingredients (APIs) or during storage of the final product [22]. Process-related impurities include byproducts, unreacted raw materials, and side products formed due to incomplete reactions or side reactions during the manufacturing process. Degradation products are formed through chemical breakdown of the API due to exposure to environmental factors such as heat, moisture, or light. HPLC is particularly effective in separating and quantifying these organic impurities, using methods such as reversed-phase chromatography and UV-Vis detection to monitor their concentration in complex formulations [28].
Inorganic impurities primarily consist of residual reagents, catalysts, and metallic contaminants. These can originate from the raw materials, reagents used in the synthesis, or leaching from manufacturing equipment. Common examples include metal traces such as iron, aluminum, or copper, which can contaminate the API during synthesis or processing. The precise quantification of these impurities often requires HPLC coupled with inductively coupled plasma (ICP) or atomic absorption spectroscopy (AAS), though HPLC itself is frequently employed in conjunction with conductivity detectors for the detection of ionic species [27].
Residual solvents are volatile organic compounds used during the manufacturing process, such as acetone, methanol, or ethanol, which must be removed or reduced to safe levels to comply with regulatory standards such as ICH Q3C [26]. These solvents, if not properly removed, can pose risks to patient health, and their detection is critical. HPLC, particularly gas chromatography (GC) or headspace analysis, is often employed for the quantification of residual solvents, ensuring that the concentration of these solvents meets the stringent regulatory limits established for pharmaceutical products [5].
In summary, the identification and quantification of these impurities, as per ICH guidelines, are essential for the quality control and safety of pharmaceutical products. HPLC plays a critical role in the separation and analysis of these impurities, ensuring compliance with pharmacopeial standards and contributing to the overall safety and efficacy of drug products.
Regulatory Framework for Method Development
Method development in High-Performance Liquid Chromatography (HPLC) is a rigorous and systematic process critical to ensuring reliable, reproducible, and efficient analytical outcomes, particularly in regulated sectors such as the pharmaceutical industry. The key to successful method development lies in selecting and optimizing both the stationary phase and mobile phase, which directly influence the separation efficiency, resolution, and retention times of analytes [28].
The stationary phase in HPLC must be chosen to provide optimal interactions with the analyte(s), facilitating selective retention and separation based on properties such as polarity, size, or charge. Reversed-phase stationary phases, such as C18, are commonly employed due to their robustness and versatility in separating a broad range of compounds. The surface characteristics and the functionalization of the stationary phase significantly impact chromatographic behaviour, affecting factors like selectivity and efficiency [29].
The mobile phase, typically a solvent or a mixture of solvents, must be carefully selected to facilitate the efficient movement of analytes through the column. Polarity, ionic strength, pH, and viscosity of the mobile phase all play pivotal roles in controlling the retention time and separation characteristics of the compounds. Tailoring the mobile phase composition enhances the interaction between the analyte and the stationary phase, optimizing chromatographic resolution and sensitivity for impurity detection [28] Gradient elution is often employed to improve the separation of complex mixtures [30].
In HPLC, temperature control is essential for method development. Although temperature has less impact on analyte volatility compared to Gas Chromatography (GC), it significantly influences viscosity, diffusion rates, and interaction kinetics between analytes and the stationary phase. Optimizing the column temperature improves peak shapes, resolution, and overall separation efficiency, especially for compounds with varying degrees of polarity or molecular weight [30].
For a method to be considered effective and reliable, it must exhibit high reproducibility and robustness across a range of analytical conditions. The method must yield consistent, accurate results under varying conditions, across laboratories, or over time. Robustness ensures minor variations in chromatographic processes, such as slight changes in temperature, mobile phase composition, or flow rate, do not unduly affect results [5].
Regulatory bodies such as the FDA, EMA, and ICH impose stringent requirements for method development, particularly for pharmaceutical applications. These agencies mandate that all HPLC methods undergo rigorous validation to confirm their accuracy, precision, specificity, linearity, and sensitivity. Validation provides assurance that methods adhere to regulatory standards and meet quality assurance protocols, ensuring reliable results for regulatory scrutiny [31]. In conclusion, method development for HPLC in the pharmaceutical industry requires a detailed, scientifically grounded approach to selecting and optimizing system components and operating conditions. Adhering to regulatory guidelines and performing rigorous validation ensures the reliability, accuracy, and compliance of analytical methods, safeguarding the quality and safety of pharmaceutical products.
Table 1: Details of method development for different drug substances.
|
Drug Substance |
HPLC Mode |
Column Details |
Detector |
Citation |
|
Paracetamol |
Reverse Phase |
C18 (150 mm × 4.6 mm, 5 µm) |
UV-Visible |
[32] |
|
Ibuprofen |
Reverse Phase |
C18 (250 mm × 4.6 mm, 5 µm) |
UV-Visible |
[33] |
|
Ciprofloxacin |
Reverse Phase |
C18 (250 mm × 4.6 mm, 5 µm) |
UV-Visible |
[34] |
|
Azithromycin |
Reverse Phase |
C18 (250 mm × 4.6 mm, 5 µm) |
UV-Visible |
[35] |
|
Ritonavir |
Reverse Phase |
C18 (250 mm × 4.6 mm, 5 µm) |
UV-Visible |
[36] |
Method Validation Guidelines in High-Performance Liquid Chromatography
Method validation is an essential process to confirm that analytical methods provide consistent, reliable, and accurate results, particularly for routine analysis in highly regulated environments such as the pharmaceutical industry. The following validation parameters are critical in ensuring that HPLC methods meet the required scientific and regulatory standards [31].
Specificity:
Specificity refers to the ability of the analytical method to distinguish the target analyte from potential interferences in a sample matrix. This ensures that only the compound of interest is measured, free from contributions from other substances that may be present in the sample. Achieving high specificity in HPLC requires the careful selection of stationary and mobile phases, along with optimization of chromatographic conditions that allow for clear separation of the analyte from potential co-eluting components [28].
Accuracy:
Accuracy is the degree to which the measured value deviates from the true or known value. In HPLC, accuracy is evaluated by comparing the results from the method with a reference or standard solution. A validated method should consistently yield results that are close to the true concentration of the analyte, ensuring that the analytical data is reliable for quantitative analysis [5].
Precision:
Precision assesses the reproducibility of the analytical method under consistent experimental conditions. It is evaluated by performing replicate analyses of the same sample and assessing the variation in results. Precision is typically quantified as the relative standard deviation (RSD) and is categorized into repeatability (within the same day) and intermediate precision (over multiple days or analysts). High precision ensures that the HPLC method produces consistent and reproducible results, which is essential for routine use in quality control laboratories [3].
Linearity:
Linearity refers to the proportionality between the analyte concentration and the detector response over a specified concentration range. A linear relationship allows for accurate quantification of varying amounts of the analyte in the sample. The method should demonstrate a correlation coefficient (r?2;) close to 1 across the expected range of concentrations, ensuring that the detector's response is uniform and reliable for quantifying both low and high levels of the analyte [37].
Limit of Detection (LOD) and Limit of Quantification (LOQ):
The LOD and LOQ are critical parameters that define the sensitivity of the HPLC method.
Robustness
Robustness evaluates the method’s ability to remain unaffected by small variations in experimental conditions, such as slight changes in temperature, mobile phase composition, or column lot. A robust method ensures reliable performance even under minor deviations from standard operating procedures, making it suitable for use in different laboratory settings and over time [3].
In conclusion, method validation for HPLC is a comprehensive and systematic process that ensures the reliability and compliance of analytical methods. By addressing critical parameters such as specificity, accuracy, precision, and robustness, validated methods support the pharmaceutical industry in delivering high-quality products that meet regulatory standards and safeguard patient health.
Table 2: Details of method validation parameters and their importance.
|
Validation Parameter |
Description |
Importance in HPLC |
Citation |
|
Specificity |
The ability of the method to distinguish the analyte from other components or interferences in the sample. |
Ensures that only the target analyte is measured, free from interference by co-eluting compounds. |
[31] |
|
Accuracy |
The degree to which the measured value is close to the true or known value. |
Confirms that the method provides reliable and correct quantitative results for the analyte of interest. |
[38] |
|
Precision |
The reproducibility of results when the same sample is analysed under the same conditions multiple times. |
Ensures consistency of results across different runs, analysts, or laboratory conditions. |
[39] |
|
Linearity |
The proportionality between analyte concentration and detector response over a defined range of concentrations. |
Ensures the method can quantify varying amounts of analyte reliably across a broad concentration range. |
[40] |
|
Limit of Detection (LOD) |
The smallest concentration of the analyte that can be detected with a reasonable degree of certainty, though not necessarily quantified. |
Determines the method’s sensitivity for detecting trace levels of an analyte. |
[41] |
|
Limit of Quantification (LOQ) |
The lowest concentration at which the analyte can be reliably quantified with acceptable precision and accuracy. |
Ensures reliable quantification of low concentrations of the analyte, necessary for detecting impurities or low-level drugs. |
[32] |
|
Robustness |
The method's ability to perform reliably under slight variations in experimental conditions, such as minor changes in temperature or mobile phase composition. |
Confirms that the method will produce consistent results even with small deviations from optimal conditions. |
[39] |
These validation parameters, as guided by regulatory standards such as those from the FDA, EMA, and ICH, ensure that the HPLC method is fit for purpose, accurate, reliable, and compliant with stringent quality control requirements. Method validation provides confidence in the analytical data produced, ensuring the safety, efficacy, and quality of pharmaceutical products.
Risk Assessments for Genotoxic Impurities (GTIs)
Effective risk assessments are vital for identifying potential sources of genotoxic impurities (GTIs) in pharmaceutical manufacturing and evaluating their impact on drug safety. These assessments aim to quantify the risks associated with GTIs and implement strategies to minimize their presence in drug products. Systematic techniques, such as Failure Mode and Effects Analysis (FMEA), are employed to identify potential failure modes within manufacturing processes and pinpoint stages where GTIs may be introduced [45]. Moreover, control threshold determinations, guided by toxicological data and regulatory limits, are utilized to establish acceptable levels of GTIs in pharmaceutical products [44]. These assessments enable the prioritization of mitigation measures, including enhancing manufacturing controls and refining purification processes, to meet the safety standards set by regulatory authorities such as the FDA and EMA [43]. Addressing potential risks at the early stages of drug development allows manufacturers to reduce the likelihood of GTIs reaching patients, thereby ensuring product safety and efficacy. A critical component of this approach is the in-silico evaluation of the genotoxicity of drug substances and raw materials. Tools like Derek Nexus, Sarah Nexus, and Lhasa are employed in accordance with the ICH M7 guideline, which focuses on the "Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk" [1]. This evaluation involves:
This integrated approach ensures that any mutagenic risks are thoroughly assessed, classified, and managed. By proactively addressing these risks, pharmaceutical companies can safeguard patients while maintaining compliance with global regulatory standards
Table 3: Impurity Classification with Respect to Mutagenic and Carcinogenic potential proposed in the ICH M7 Guidance.
|
Class |
Definition |
Proposed action for control |
Citation |
|
1 |
Known mutagenic carcinogens |
Control at or below compound-specific acceptable limit |
[46] |
|
2 |
Known mutagens with unknown carcinogenic potential (Bacterial mutagenicity positive), no rodent carcinogenicity data) |
Control at or below acceptable limits (Appropriate TTC) |
[47] |
|
3 |
Alerting structure, unrelated to the structure of the drug substance; no mutagenicity data |
Control at or below acceptable limits (Appropriate TTC) or conduct bacterial mutagenicity assay. If non-mutagenic = Class 5 If mutagenic = Class 2 |
[48] |
|
4 |
Alerting structure, same alert in drug substance or compounds related to the drug substance (e.g., process intermediates) which have been tested and are non-mutagenic |
Treat as non-mutagenic impurity |
[49] |
|
5 |
No structural alerts, or alerting structure with sufficient data to demonstrate lack of mutagenicity or carcinogenicity |
Treat as non-mutagenic impurity |
[50] |
Limit Calculation:
Formula:
1.5 µg
Calculation (ppm) = --------------------------------
Maximum daily dosage of drug substance in grams
Control Strategies for Genotoxic Impurities (GTIs)
Control strategies are critical to maintaining genotoxic impurities (GTIs) in pharmaceutical products within acceptable limits, ensuring compliance with regulatory standards. These strategies involve a combination of analytical controls, process optimization, and adherence to regulatory guidelines:
Regular testing and monitoring are employed throughout the manufacturing process to detect and quantify impurities, ensuring they remain below established thresholds. Techniques such as gas chromatography (GC) and high-performance liquid chromatography (HPLC) are commonly used to identify and measure GTIs with high precision and sensitivity 51].
Manufacturing processes are optimized to minimize the formation of GTIs. This includes refining synthesis conditions, improving purification methods, and implementing advanced separation technologies to reduce impurity levels effectively. These measures not only enhance product safety but also improve process efficiency [44].
Ensuring adherence to safety standards, such as those outlined in the ICH M7 guidelines, is a fundamental aspect of control strategies. These guidelines provide a framework for the assessment and control of mutagenic impurities, emphasizing the importance of risk-based approaches and threshold of toxicological concern (TTC) principles [1].
Table 4: The control of mutagenic and carcinogenic impurities according to the ICH M7 Guidance
|
Product Name |
Impurity Name |
ICH M7 Classification |
Limit |
LOQ (ppm) |
LOD (ppm) |
Citation |
|
Paracetamol |
4-Aminophenol |
Genotoxic |
1.5 ppm |
0.15 |
0.05 |
[52] |
|
Ibuprofen |
Isobutyl benzene |
Genotoxic |
2 ppm |
0.1 |
0.03 |
[53] |
|
Ciprofloxacin |
Piperazine derivative |
Genotoxic |
2 ppm |
0.08 |
0.02 |
[54] |
|
Azithromycin |
Aziridine |
Genotoxic |
1.0 ppm |
0.05 |
0.01 |
[55] |
|
Ritonavir |
Thioamide derivative |
Genotoxic |
2 ppm |
0.07 |
0.02 |
[56] |
CONCLUSION
HPLC continues to serve as a gold-standard analytical technique for the detection and quantification of genotoxic impurities (GTIs) in pharmaceuticals. Ongoing advancements in technology and evolving regulatory frameworks have further enhanced its precision, sensitivity, and applicability. Emerging innovations, such as automation and eco-friendly "green" chromatography techniques, are poised to expand HPLC’s capabilities, reinforcing its pivotal role in safeguarding the safety and efficacy of pharmaceutical products.
REFERENCES

M. Manivardhan Reddy*, G. Sampath Kumar Reddy, and K. Ramadevi, Comprehensive Insights into High-Performance Liquid Chromatography for Pharmaceutical Analysis: Focus on Genotoxic Impurities, Int. J. of Pharm. Sci., 2025, Vol 3, Issue 1, 2031-2046. https://doi.org/10.5281/zenodo.14729254
 10.5281/zenodo.14729254
10.5281/zenodo.14729254