
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1. Research Scholar, IEC School of Pharmacy, IEC University, Kalujhanda, Baddi, Solan, Himachal Pradesh, 174103, India
2. Associate Professor, IEC School of Pharmacy, IEC University, Kalujhanda, Baddi, Solan, Himachal Pradesh, 174103, India
3. Head of Department, IEC School of Pharmacy, IEC University, Kalujhanda, Baddi, Solan, Himachal Pradesh, 174103, India
Computational chemistry and artificial intelligence (AI) are rapidly reshaping the landscape of pharmaceutical research and drug development. Conventional drug discovery methods are often expensive, time-intensive, and associated with high attrition rates during preclinical and clinical studies. The application of computational techniques such as molecular docking, molecular dynamics simulations, pharmacophore modelling, virtual screening, and quantitative structure–activity relationship (QSAR) analysis has greatly improved the ability to understand molecular interactions and optimize drug candidates. In parallel, AI-based technologies including machine learning, deep learning, graph neural networks, and generative models have enhanced the prediction of biological activity, toxicity, pharmacokinetic behaviour, and drug–target interactions. These advanced computational approaches facilitate faster target identification, lead optimization, de novo molecular design, and drug repurposing. Additionally, the integration of big data analytics, bioinformatics, natural language processing, and cloud computing has strengthened data-driven pharmaceutical research. AI-assisted systems are increasingly being implemented throughout the drug discovery pipeline, from early-stage screening to clinical trial optimization and personalized medicine. Despite these advancements, several challenges remain, including limited data quality, model transparency, regulatory concerns, and the need for experimental validation.This review summarizes recent developments in computational chemistry and AI-assisted drug discovery, highlighting important technologies, applications, current limitations, and future opportunities in next-generation pharmaceutical innovation.
Drug discovery is a complex and time-consuming process requiring substantial financial investment and scientific effort. Computational chemistry and AI have emerged as transformative technologies capable of accelerating drug development while improving prediction accuracy and reducing failure rates.
Computational Chemistry in Drug Discovery
Computational chemistry techniques such as molecular docking, molecular dynamics simulations, quantum mechanical calculations, and pharmacophore modelling provide molecular-level understanding of drug-target interactions. These methods assist in structure-based and ligand-based drug design.
The integration of computational chemistry with AI has led to major improvements in structure-based drug design, ligand-based drug discovery, toxicity prediction, and drug repurposing.”
The overall workflow of AI-assisted drug discovery is illustrated in Figure 1.
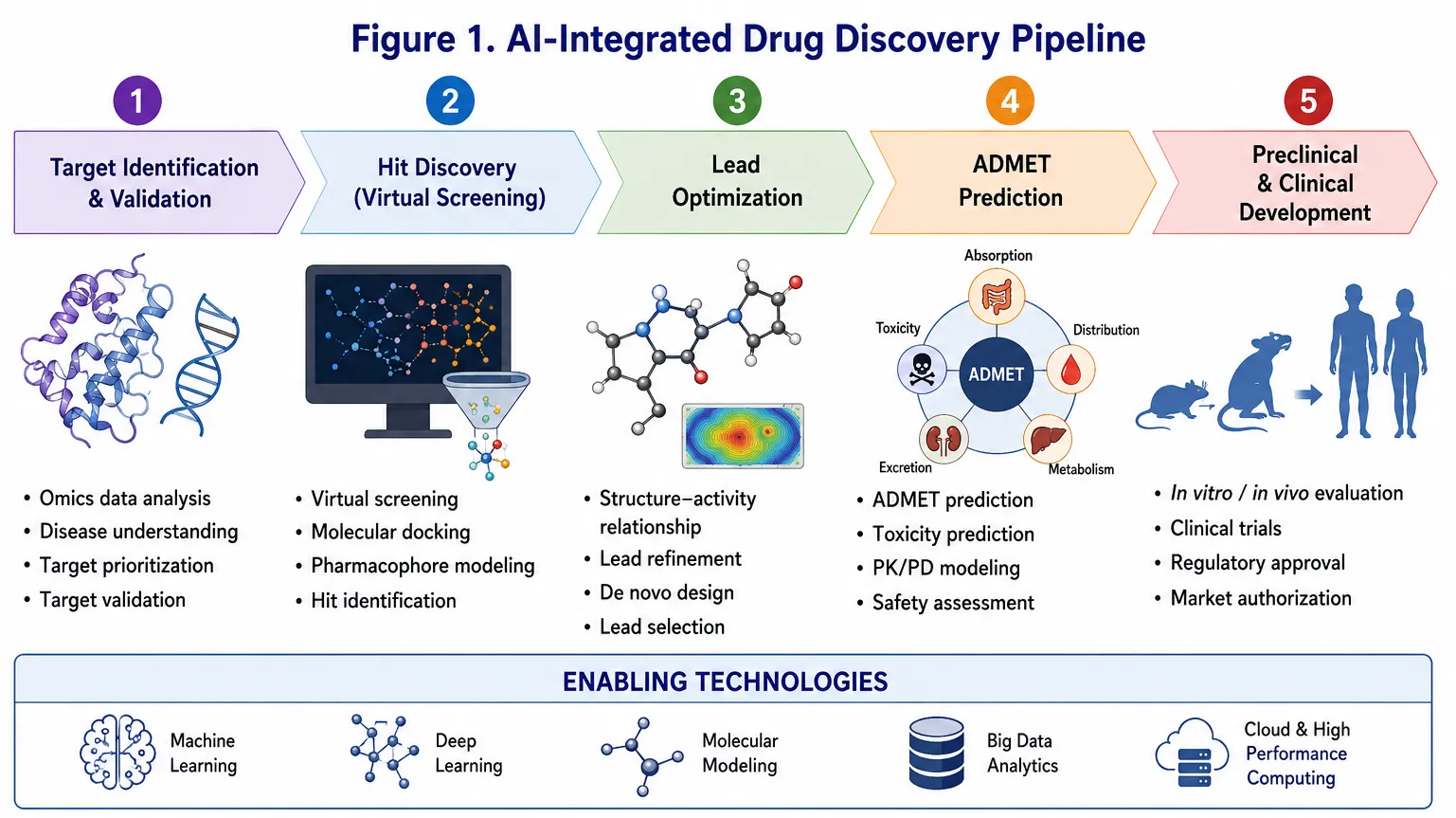
Figure 1. Overview of the AI-integrated drug discovery pipeline showing major stages including target identification, virtual screening, lead optimization, ADMET prediction, and preclinical/clinical development.
Artificial Intelligence in Drug Discovery
Machine learning and deep learning models are increasingly applied in target identification, virtual screening, toxicity prediction, lead optimization, and personalized medicine. AI models can analyse massive biological datasets and identify hidden relationships beyond human capability.
AI-Driven Target Identification and Validation
AI-assisted systems integrate genomics, proteomics, transcriptomics, and network biology to identify disease-associated targets and prioritize therapeutically relevant proteins for drug development.
Drug Repurposing and Polypharmacology
Drug repurposing uses existing approved drugs for new therapeutic applications, significantly reducing development time and cost. Polypharmacology focuses on designing drugs that interact with multiple biological targets.
AI in ADMET Prediction and Toxicity Assessment
AI-based toxicity prediction platforms can identify hepatotoxicity, cardiotoxicity, mutagenicity, and other safety concerns early during drug development, reducing late-stage failures.
Integration of AI into the Drug Discovery Pipeline
AI is increasingly integrated into every stage of pharmaceutical development, including target selection, lead optimization, clinical trial design, and pharmacovigilance..
Challenges and Limitations
Challenges include poor data quality, model interpretability issues, regulatory uncertainty, ethical concerns, and the need for experimental validation of computational predictions.
Future Perspectives
Future drug discovery will likely involve AI-guided personalized medicine, autonomous robotic laboratories, quantum computing, and real-time pharmacovigilance systems.
Role of Big Data in Drug Discovery
The pharmaceutical industry generates enormous amounts of biological, chemical, genomic, proteomic, and clinical data. Big data analytics enables researchers to process and interpret these datasets efficiently. AI algorithms can identify hidden relationships between genes, proteins, diseases, and drug molecules, thereby improving target identification and biomarker discovery.
Integration of electronic health records (EHRs), clinical trial databases, and real-world evidence has further enhanced predictive modelling and personalized medicine. Big data platforms also support rapid drug repurposing by identifying unexpected therapeutic relationships between approved drugs and diseases.
Applications of Big Data
Role of Quantum Computing in Drug Discovery
Quantum computing has emerged as a promising technology for solving highly complex molecular calculations that are difficult for classical computers. Quantum algorithms can improve molecular simulations, protein folding predictions, and quantum mechanical calculations involved in drug design.
Although still in early stages, quantum computing may revolutionize computational chemistry by enabling highly accurate simulations of molecular interactions and chemical reactions. Integration of quantum computing with AI may substantially accelerate lead optimization and molecular property prediction.
Potential Benefits
Personalized Medicine and AI
Personalized medicine aims to provide treatments tailored to an individual’s genetic makeup, lifestyle, and disease profile. AI-driven systems analyse patient-specific genomic and clinical data to predict drug response and optimize therapeutic strategies.
Machine learning algorithms can identify biomarkers associated with treatment outcomes, helping clinicians select the most effective drugs while minimizing adverse effects. AI-assisted precision medicine is especially valuable in oncology, neurology, and rare genetic diseases.
Advantages
Role of Natural Language Processing in Drug Discovery
Natural Language Processing (NLP) is increasingly used to analyse scientific literature, patents, clinical reports, and biomedical databases. NLP systems can rapidly extract meaningful information from millions of published articles and identify relationships between diseases, genes, proteins, and drugs.
AI-powered NLP tools assist in:
Large language models (LLMs) are also emerging as powerful tools for biomedical knowledge synthesis and hypothesis generation.
AI in Clinical Trials
Clinical trials are one of the most expensive and time-consuming stages of drug development. AI technologies improve clinical trial efficiency through:
AI-driven patient stratification helps identify suitable patient populations, improving the probability of clinical success while reducing trial costs.
Regulatory Perspectives on AI-Based Drug Discovery
Regulatory agencies such as the U.S. Food and Drug Administration and European Medicines Agency are actively developing frameworks for evaluating AI-assisted pharmaceutical development.
Key regulatory concerns include:
Future regulatory guidelines are expected to encourage responsible adoption of AI while ensuring patient safety and scientific reliability.
Case Studies of AI-Assisted Drug Discovery
AI in COVID-19 Drug Discovery
During the COVID-19 pandemic, AI and computational chemistry played major roles in identifying antiviral compounds, predicting protein structures, and repurposing approved drugs. Virtual screening and molecular docking were widely used to identify inhibitors targeting SARS-CoV-2 proteins.
AI-Designed Drug Candidates
Several AI-designed molecules have entered preclinical and early clinical development. AI-driven platforms have demonstrated the ability to shorten lead identification timelines from years to months.
These case studies highlight the growing practical impact of AI in real-world pharmaceutical research.
Industrial Adoption of AI in Pharmaceutical Research
Major pharmaceutical companies are increasingly collaborating with AI-focused biotechnology firms to improve R&D productivity. AI-driven platforms are now widely used in:
The global market for AI in drug discovery is expected to grow substantially over the coming decade due to increased investment and technological advancement.
Ethical Considerations in AI-Driven Drug Discovery
Although AI offers numerous advantages, ethical challenges remain important.
Major ethical concerns include:
Responsible AI governance and human oversight are essential for maintaining scientific integrity and patient trust.
Final Future Outlook
The future of drug discovery will likely involve fully integrated AI-driven pharmaceutical ecosystems combining:
These technologies may eventually enable continuous, adaptive, and personalized therapeutic development with dramatically reduced timelines and costs.
CONCLUSION
Computational chemistry and AI are revolutionizing modern pharmaceutical research by accelerating drug discovery, improving prediction accuracy, and reducing development costs. These technologies are expected to become central components of future drug development strategies.
REFERENCES


Mohamed Abdulla Mohammad Abdulla , Shalini Devi, Sunita Dhiman, Swati Joshi, Jyoti Gupta, Recent Progress in Drug Discovery Using Computational Chemistry and Artificial Intelligence, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 8356- 8365. https://doi.org/ 10.5281/zenodo.20477294
 10.5281/zenodo.20477294
10.5281/zenodo.20477294