
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutics, Kamalakshi Pandurangan College of Pharmacy, Ayyampalayam, Tiruvannamalai – 606 603
Post-approval changes in manufacturing site transfer represent one of the most critical lifecycle management activities in pharmaceutical regulation. The transfer of a manufacturing site for an approved drug product requires regulatory oversight to ensure that product quality, safety, and efficacy remain unaffected. The United States Food and Drug Administration (US FDA) regulate such changes through a structured framework based on risk categorization, prior approval supplements, and current Good Manufacturing Practices (cGMP). Over the past two decades, regulatory expectations have evolved significantly, influenced by science- and risk-based approaches introduced through International Council for Harmonisation (ICH) guidelines and FDA modernization initiatives. This review provides a detailed analysis of regulatory requirements governing site transfer approvals in the United States, examines the evolution of policy, discusses submission pathways, comparability strategies, inspectional expectations, and industry challenges, and highlights future directions in regulatory science. The article synthesizes regulatory guidance documents, published literature, and inspectional trends to provide a comprehensive scholarly review suitable for academic and professional audiences.
The globalization of pharmaceutical manufacturing has transformed the operational landscape of drug production. Pharmaceutical companies frequently relocate manufacturing activities due to capacity expansion, cost optimization, mergers and acquisitions, supply chain resilience strategies, or compliance remediation. Such relocation, commonly termed site transfer, involves moving manufacturing operations of an approved drug product from one facility to another. While business-driven, this process is heavily regulated because any change in facility may potentially impact product quality attributes. (1)
The regulatory oversight of site transfer changes in the United States is governed by the statutory framework of the Federal Food, Drug, and Cosmetic Act and implementing regulations within Title 21 of the Code of Federal Regulations (CFR). The U.S. Food and Drug Administration (FDA) evaluates post-approval site changes using risk-based classification systems to determine the appropriate reporting category. The primary objective is to ensure that transferred manufacturing operations continue to produce drug products meeting established specifications without introducing variability that may compromise patient safety.
This review aims to analyze the regulatory mechanisms governing site transfer approvals, examine historical and contemporary guidance documents, discuss risk-based evaluation approaches, and assess practical challenges encountered during regulatory submissions and inspections. (2)
2. Regulatory Framework Governing Site Transfer
The regulatory control of site transfer is anchored in 21 CFR Parts 210 and 211, which establish Current Good Manufacturing Practices (cGMP) requirements for finished pharmaceuticals. These regulations require that manufacturing facilities maintain validated processes, qualified equipment, trained personnel, and adequate quality systems. Any change in the manufacturing site is considered a significant post-approval modification because environmental conditions, utilities, equipment configuration, and operational practices may differ between facilities.
Post-approval changes are formally categorized under supplement reporting regulations codified in 21 CFR 314.70 for New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs). Depending on the potential risk to product quality, site transfers may require submission as a Prior Approval Supplement (PAS), a Changes Being Effected (CBE-30), or inclusion in an Annual Report. Generally, a complete transfer of manufacturing to a new facility not previously approved in the application requires a PAS because it is considered to have substantial potential to adversely affect identity, strength, quality, purity, or potency.
The FDA’s approach emphasizes that regulatory reporting category is determined not solely by physical relocation but by the risk posed to product quality attributes. This approach aligns with modern regulatory philosophy centered on science- and risk-based decision making.
3. Regulatory Pathway Flow Diagram
The regulatory approval pathway of site transfer was showed in the Figure 1.
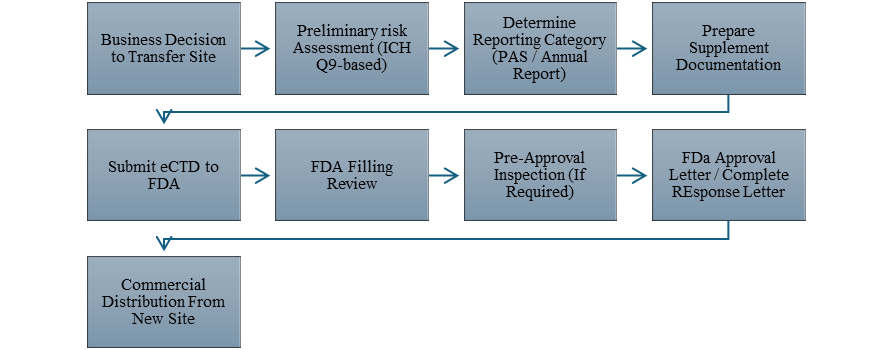
Figure 1. FDA Site Transfer Approval Pathway
4. Evolution of FDA Policy on Site Transfer
The regulatory approach to post-approval manufacturing changes evolved significantly following the FDA’s pharmaceutical cGMP modernization initiative in 2004. Historically, regulatory review was largely compliance-oriented and prescriptive. However, integration of quality systems thinking and risk management principles transformed regulatory oversight into a more flexible and scientifically grounded system.
The introduction of ICH quality guidelines profoundly influenced FDA expectations. ICH of Technical Requirements forS Pharmaceuticals for Human Use developed harmonized frameworks that underpin modern pharmaceutical development and lifecycle management. The adoption of ICH Q8 introduced the concept of design space and systematic pharmaceutical development. ICH Q9 established formal quality risk management methodologies, while ICH Q10 emphasized lifecycle quality systems and change management. (4)
These guidelines collectively shifted regulatory evaluation of site transfers from a documentation-driven exercise to a structured comparability assessment based on risk evaluation, process understanding, and quality system robustness.
5. Business Decision to Transfer Manufacturing Site
The first step in site transfer begins with a strategic business decision by the pharmaceutical company to relocate manufacturing activities. This decision may be driven by multiple operational or commercial factors. (5)
Common reasons for site transfer
Before implementing the change, the company must evaluate the regulatory impact because a change in manufacturing site can potentially affect:
Therefore, the proposed change must be formally initiated through the change control system of the pharmaceutical quality management system described in ICH Q10. (7)
6. Risk Assessment
After initiating change control, a comprehensive risk assessment is conducted to determine the potential impact of transferring manufacturing operations to a new facility.
Risk evaluation follows the principles of ICH Q9, which recommends systematic identification, evaluation, and control of risks affecting product quality. (8)
Key factors assessed during risk evaluation
The table 1 shown that the risk factor and its potential impact: -
Table 1. Key factors assessed during risk evaluation
|
Risk Factor |
Potential Impact |
|
Equipment differences |
Changes in process parameters |
|
Facility design |
Environmental control variations |
|
Personnel training |
Operational variability |
|
Utilities (HVAC, water systems) |
Stability and contamination risks |
|
Raw material supply chain |
Quality variation |
Risk assessment tools commonly used
The outcome of this stage determines whether additional validation studies, comparability studies, or regulatory supplements are required. (9)
7. Determination of Regulatory Reporting Category
Following risk assessment, the regulatory affairs team determines the appropriate reporting category according to 21 CFR 314.70.
Regulatory categories for site transfer
Types of changes in site transfer was shown in the table 2.
Table 2. Types of changes in site transfer
|
Change Category |
Submission Type |
Description |
|
Major Change |
Prior Approval Supplement (PAS) |
Requires FDA approval before product distribution |
|
Moderate Change |
Changes Being Effected in 30 Days (CBE-30) |
Product may be distributed after 30 days if FDA raises no objection |
|
Minor Change |
Annual Report |
Low-risk changes reported annually |
Site transfers typically fall under PAS, especially when the new manufacturing site has not been previously approved for the product.
Additional regulatory guidance related to manufacturing changes is provided in SUPAC, which describes acceptable manufacturing modifications after approval. (10)
8. Preparation of Regulatory Submission (eCTD)
Once the reporting category is determined, the company prepares a regulatory submission using the Electronic Common Technical Document (eCTD) format defined by ICH M4 Common Technical Document.
Most site transfer documentation is included in Module 3 (Quality) of the CTD dossier.
Key documentation required
Table 3. Key documentation required for regulatory submission
|
CTD Section |
Information Provided |
|
Cover Letter |
Description of the proposed change |
|
FDA Form 356h |
Application amendment |
|
Module 3.2.S |
Drug substance manufacturing details |
|
Module 3.2.P |
Drug product manufacturing site details |
|
Process Validation Reports |
Validation batch results |
|
Analytical Method Validation |
QC method transfer data |
|
Stability Data |
Accelerated and long-term stability |
|
Environmental Assessment |
Required under 21 CFR 25 |
Additional documentation may include technology transfer reports, equipment qualification records (IQ/OQ/PQ), and comparability data. (11) The key documentation required for regulatory submission was represented in the tble 3.
9. FDA Filing Review
After submission, the dossier undergoes a filing review by the U.S. Food and Drug Administration to determine whether the application contains sufficient information for detailed evaluation.
During this stage, the agency verifies:
If the submission is considered complete, it proceeds to scientific review by FDA experts in chemistry, manufacturing, and controls (CMC). (12)
10. Pre-Approval Inspection (PAI)
In many cases, the FDA conducts a Pre-Approval Inspection (PAI) of the new manufacturing facility before granting approval.
The objective of the PAI is to confirm that the facility complies with current Good Manufacturing Practice regulations under 21 CFR Parts 210 and 211.
Inspection areas typically evaluated
If significant deficiencies are identified, the FDA may issue a Form 483 observation requiring corrective actions. (13)
11. FDA Approval or Complete Response Letter
Following scientific review and inspection, the FDA issues one of the following regulatory decisions:
Approval
If all regulatory requirements are satisfied, the FDA grants approval for the site transfer, allowing commercial manufacturing at the new facility.
Complete Response Letter (CRL)
If deficiencies remain, the FDA issues a Complete Response Letter, requesting additional information, corrective actions, or further studies before approval can be granted. (14)
12. Commercial Production at the New Site
After approval, the company may begin commercial production at the new manufacturing site. However, the manufacturer must continue monitoring product quality through:
These lifecycle quality management practices align with recommendations in ICH Q12. (15)
13. Detailed Documentation Requirements
A complete site transfer supplement typically includes:
13.1 Administrative Documentation
13.2 Quality Documentation
13.3 Analytical Documentation
13.4 Risk Management Documentation
14. Comparability Study Design
Comparability ensures no adverse impact on product quality after transfer.
14.1 Small Molecule Products
14.2 Biologics
15. Reporting Categories and Submission Pathways
The classification of site transfer changes is guided by the FDA’s 1997 Guidance for Industry on Changes to an Approved NDA or ANDA and subsequent updates. A Prior Approval Supplement is required when a site change presents substantial potential risk. Examples include transfer of drug substance manufacturing to a new facility, relocation of sterile manufacturing operations, or transfer involving complex biologics production.
Moderate-risk transfers may be eligible for CBE-30 reporting if sufficient validation data demonstrate equivalence and the facility is already compliant with FDA requirements. Minor changes may be reported in the Annual Report if they pose minimal risk and do not affect critical process parameters or product specifications. (19)
The decision matrix integrates risk assessment, process validation status, facility inspection history, and product complexity. Sterile products, biologics, and modified-release formulations are subject to heightened scrutiny due to higher clinical risk associated with variability. (20)
16. Comparability Assessment in Site Transfers
A core regulatory expectation in site transfer is demonstration of comparability between pre-transfer and post-transfer product. Comparability assessment encompasses analytical testing, process validation, stability studies, and sometimes clinical bridging if major process modifications accompany relocation.
For small-molecule products, demonstration of equivalent impurity profiles, dissolution characteristics, assay results, and stability performance is typically sufficient. For biologics, comparability exercises are more complex, involving characterization of higher-order structure, glycosylation patterns, biological activity, and immunogenicity risk assessment.
The FDA encourages proactive use of comparability protocols, which outline pre-approved testing strategies and acceptance criteria. When properly designed, these protocols facilitate streamlined regulatory review and reduce uncertainty. (21) An example of comparability testing matrix was shown in the table 4.
Table 4. Comparability Testing Matrix
|
Parameter |
Pre-Transfer Data |
Post-Transfer Data |
Acceptance Criteria |
|
Assay |
99.5% |
99.3% |
98–102% |
|
Dissolution |
Q=85% |
Q=87% |
NLT 80% |
|
Impurity A |
0.15% |
0.16% |
NMT 0.5% |
17. Risk Classification and Reporting Categories
Site transfer changes are categorized according to potential risk to product quality that was represented in the table 5. (22)
Table 5. FDA Reporting Categories for Site Transfer
|
Reporting Category |
Risk Level |
Examples |
FDA Review Timeline |
|
Prior Approval Supplement (PAS) |
High |
New sterile facility, biologics manufacturing transfer |
6–10 months |
|
CBE-30 |
Moderate |
Solid oral dosage transfer to compliant facility |
30 days |
|
CBE-0 |
Moderate–Low |
Certain labeling-related site updates |
Immediate |
|
Annual Report |
Low |
Minor warehouse relocation |
Annual reporting cycle |
18. Inspectional Considerations
Manufacturing site transfers frequently trigger FDA pre-approval inspections to verify readiness of the new facility. Inspectional focus areas include process validation documentation, equipment qualification, cleaning validation, data integrity systems, training records, and quality oversight mechanisms.
Analysis of publicly available warning letters indicates recurring deficiencies in inadequate process validation, insufficient comparability data, and incomplete stability commitments. Facilities with robust quality management systems aligned with ICH Q10 principles demonstrate smoother approval outcomes. (23)
19. Industry Challenges and Operational Impact
Industry faces several operational challenges during site transfers. These include coordination between sending and receiving units, alignment of documentation systems, validation scheduling, and management of supply continuity. Global harmonization adds complexity when products are registered in multiple jurisdictions with differing regulatory expectations.
Delays in approval of site transfer supplements can lead to drug shortages, particularly when the original facility ceases operations before approval of the new site. Regulatory agencies have increasingly emphasized early communication to mitigate such risks. (24)
12. Impact of Regulatory Modernization
The FDA’s modernization initiatives, including Quality Metrics programs and risk-based inspection models, have enhanced transparency and predictability in post-approval change management. Digital submission platforms and structured electronic Common Technical Document (eCTD) formats have improved review efficiency.
Regulatory reliance and mutual recognition agreements between global agencies further influence site transfer strategies, although independent FDA approval remains mandatory for U.S. market authorization. (25)
20. Future Perspectives
Emerging technologies such as continuous manufacturing, advanced analytics, and digital twins are expected to influence site transfer strategies. Increased adoption of predictive quality analytics may allow more streamlined post-approval change reporting categories. Regulatory convergence through ICH updates may further harmonize global approaches.
The FDA continues to refine lifecycle management policies to balance innovation facilitation with patient protection. Emphasis on science-based comparability and risk management will remain central to regulatory evaluation of manufacturing site changes.
CONCLUSION
Post-approval site transfer under the USFDA framework represents a highly regulated lifecycle activity requiring integration of regulatory science, pharmaceutical technology, risk management, and quality systems. The transfer process extends beyond simple geographic relocation and demands scientific demonstration of process equivalency and product comparability.
For small-molecule drugs regulated under 21 CFR 314, reporting classification depends on the impact potential of the change. For biologics regulated under 21 CFR 601, comparability requirements are significantly more rigorous due to molecular complexity and immunogenicity considerations.
Effective site transfer requires structured documentation, validated process controls, stability assurance, and proactive inspection readiness. The increasing regulatory emphasis on lifecycle management, ICH Q12 implementation, and digital quality systems reflects a shift toward science- and risk-based regulatory oversight.
Strategic planning, early risk assessment, and robust comparability protocols are essential to prevent supply disruptions and regulatory delays. As global supply chains become increasingly complex, site transfer management will continue to remain a critical competency within pharmaceutical regulatory affairs.
ACKNOWLEDGEMENT
Express my profound gratitude to Dr. M. Bharathi, our guide, for her constant guidance, encouragement and insightful suggestions throughout the completion of this article “Post-approval changes in site transfer under the U.S. FDA regulatory framework: A comprehensive review”
REFERENCES









R. Pavithra, T. Arasu, K. Mahendiran, K. Gilbert Tony, Dr. M. Bharathi, V. Kannabirran, K. Senthilkumar, D. Rajalingam, N. Gnanasekar, Post-Approval Changes in Site Transfer Under the U.S. FDA Regulatory Framework: A Comprehensive Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 4, 76-85, https://doi.org/10.5281/zenodo.19369238
 10.5281/zenodo.19369238
10.5281/zenodo.19369238