
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
R. G. Sapkal college of Pharmacy, Anjeneri, Nashik
Chronic inflammation is a hallmark of various diseases, including autoimmune disorders, cancer, and neurodegenerative diseases. Recent advances in understanding the molecular mechanisms underlying inflammation have led to the development of novel therapeutic strategies targeting key signaling pathways, including NF-KB, NLRP3, JAK/STAT, and MAPK. This review aims to provide an overview of the recent advances in these molecular pathways and their potential as therapeutic targets for anti-inflammatory agents.
Chronic inflammation is a complex process involving multiple cell types, cytokines, and signaling pathways. The NF-KB, NLRP3, JAK/STAT, and MAPK pathways play crucial roles in regulating inflammation and immune responses. Dysregulation of these pathways has been implicated in various diseases, including autoimmune disorders, cancer, and neurodegenerative diseases.
The immune system depends on innate immunity and adaptive immunity to protect the host against stress stimuli and infectious agents. Innate immunity serves as the first line of host response, and functions in a quick and non-specific manner. It is activated by stereotypical pathogen-associated molecular patterns (PAMPs) and damage- associated molecular patterns (DAMPs), which are detected by host pattern recognition receptors (PRRs). Adaptive immunity is an antigen-specific and cell/humoral-mediated host response that encompasses four types of T cell response (antigen recognition, immune checkpoint, cytokine stimulation, and MADS recognition) and two types of B cell response (T cell-dependent and T cell-independent). In adaptive immunity, metabolite-associated danger signals (MADS) refer to small molecules derived from metabolic pathways that act as immunological cues, modulating the initiation, activation, or regulation of adaptive immune responses. These signals often arise during cellular stress, damage, or infection and influence immune cell function (e.g., T cells, B cells, antigen-presenting cells), bridging metabolic states with immune regulation.
PHARMACOLOGY OF ANTI-INFLAMMATORY AGENTS
Anti-inflammatory agents can be broadly classified into several categories, including non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, and biologic agents. Recent advances in understanding the molecular mechanisms underlying inflammation have led to the development of novel therapeutic strategies targeting specific signaling pathways.
NF-KB Pathway
The NF-KB pathway is a key regulator of pro-inflammatory gene expression. Activation of the NF-KB pathway leads to the transcription of genes involved in inflammation, immune response, and cell survival. NF-KB signaling is tightly regulated by various mechanisms, including phosphorylation, ubiquitination, and degradation of key signaling molecules.
As a regulator of pro-inflammatory cytokines, chemokines, and cell survival, NF-κB remains a key target, though its dual role in cell survival requires careful inhibition to avoid toxicity.
Physiological roles of NF-κB signaling
NF-κB plays a key role in cellular responses to external stimuli such as cytokines, stress, UV light, antigens, and heavy metals. Existing studies have demonstrated that the NF-κB signaling is involved in a diverse array of physiological and pathological processes, including immune and inflammatory responses, cell survival and proliferation, metabolism, as well as synaptic plasticity and memory-related activities (Fig. 3a).

NF-κB signaling is particularly important in regulating cellular adaptation to environmental changes. In response to inflammatory stimuli, immune cells reconfigure metabolism through cellular responses mediated by NF-κB signaling. Drosophila studies revealed that NF-κB maintains the coordination of innate immune-metabolic responses by inhibiting Foxo-mediated lipolysis. Muscle contraction involves activation of NF-κB signaling by Ca2+, peroxides, and nitrogen oxides. It has been demonstrated that NF-κB signaling is activated during the strenuous exercise of the organism, either in normoxia or acute hypoxia, which includes the increase of p105, p50, IKKα, IκBβ, and glutathione reductase protein levels as well as CaMKII δD phosphorylation. When exercise ends and the muscle resumes open circulation, these changes return. The design of the new study needs to take into account the rapid changes in NF-κB signaling during exercise cessation.
TLR-induced NF-κB activation upregulates the transcription of genes encoding inflammatory vesicles and initiates immune responses. Inflammation serves as a pivotal defense mechanism against bacterial and viral infections. Serine/threonine kinase 4 (Stk4) and NF-κB are involved in the activation and homeostasis of regulatory T (Treg) cells and promote Treg cell-mediated immune tolerance. Deletion of Stk4 in mouse Treg cells inhibits p65 expression, p65-Foxp3 complex formation, and Treg cell activation, ultimately leading to autoimmune lymphoproliferative disorders. The IKK complex protects mature T cells from TNF-induced cell death and is important for their normal homeostasis and function. The integrity of cellular function requires rapid activation and termination of NF-κB signaling, and this tight regulation is essential for normal cellular and organismal homeostasis. N6-methyladenosine (m6A) mRNA modification is involved in the maintenance of colonic epithelial cells and stem cell homeostasis. Studies in mouse colon epithelial cells have revealed that methyltransferase 14 (Mettl14) inhibits colonic epithelial cell apoptosis by modulating the NF-κB pathway.
Pathological roles of NF-κB signaling
The pathological effects of NF-κB signaling include immune disorders, malignant behavior of tumor cells, metabolic dysregulation, and skeletal disorders. These effects are further described below.
Due to the key regulatory role of NF-κB signaling in immune and inflammatory responses, its dysregulation has been strongly associated with a variety of human diseases, including cancer, inflammatory diseases, autoimmune disorders, viral infections, and infectious shock. During inflammation, the NF-κB signaling is hyperactivated, leading to the abundant expression of inflammation-associated genes. Initially, researchers discovered that NF-κB potentially contributes to the pathogenesis of acquired immune deficiency syndrome (AIDS) by synergizing with and stimulating the transcription of human immunodeficiency virus (HIV). Research on p50-deficient mice has demonstrated the crucial involvement of NF-κB in both specific and non-specific immune responses, and although there is no evidence for the involvement of NF-κB in the developmental process.
As a chronic ailment, the prevalence and fatality of neoplasms persistently escalate, posing a significant peril to human existence and well-being. NF-κB signaling is involved in tumorigenesis, progression, EMT, tumor metastasis, and drug resistance. NF-κB signaling is a major pathway mediating the interaction between inflammation and cancer. As a result of alterations in the inflammatory microenvironment and oncogenic mutations, sustained NF-κB activation and dysregulation of cellular functions are observed in cancer, leading to genomic instability and gene mutations, creating a microenvironment that promotes tumor progression and promotes proliferation and angiogenesis of tumor cells while inhibiting their apoptosis.
Recent research has unveiled the pivotal role of NF-κB in the cellular response of tumors to nutrient-deprived microenvironments, and the main mechanism is to remodel the local metabolism by coordinating the actions of glycolysis, glutaminolysis, and oxidative phosphorylation pathways. Impairment of canonical and non-canonical NF-κB signaling may lead to specific developmental and immune deficiencies. For instance, germline mutations in NFKB2 in non-canonical NF-κB signaling affect the nuclear translocation of p52, which is thought to be the genetic cause of primary immunodeficiency syndromes.
The non-canonical NF-κB pathway is crucial in lymphoid organ development, lymphocyte survival and homeostasis, dendritic cell activation, osteoclastogenesis, etc., and its aberrant activation may lead to rheumatoid arthritis, ulcerative colitis, osteoporosis, and lymphoid malignancies. Expression of RelB subunits is associated with the differentiation of dendritic cells and thymic UEA-1+ medullary epithelial cells, which provides the basis for its involvement in immune responses. NF-κB receptor activator ligand (RANKL), an osteoclast differentiation factor, has an influential role in osteoclastogenesis, linking the activated immune system to bone loss.
NLRP3 Inflammasome Pathway
The NLRP3 inflammasome is a multiprotein complex that plays a critical role in the regulation of inflammation. Activation of the NLRP3 inflammasome leads to the cleavage of pro-inflammatory cytokines, such as IL-1β and IL-18, and the induction of pyroptosis.
The NLRP3 inflammasome has emerged as a central therapeutic target for chronic inflammatory diseases, as its activation by DAMPs/PAMPs leads to IL-1β/IL-18 release and cytokine storms.
Activation of the NLRP3 inflammasome signaling pathway requires two independent and concomitant signals: signal 1 (priming) and signal 2 (activation). The priming step can be induced by Toll-like receptors (TLRs, such as TLR2 and TLR4), NLRs (such as NOD1 and NOD2), cytokine receptors [such as IL-1 receptor and tumor necrosis factor-α (TNF-α) receptor], or myeloid differentiation primary response 88 (MyD88). The activation step can be induced by PAMPs (such as nigericin, viral RNA, and MDP), DAMPs [such as extracellular ATP, mitochondrial DNA (mtDNA), and mitochondrial reactive oxygen species (mtROS)], self-derived particulates [such as uric acid, amyloid-β (Aβ), calcium crystals, and cholesterol crystals], or foreign-derived particulates (such as aluminum salts, silica crystals, and asbestos).(p16),(p17) Nuclear receptors (NRs) regulate immunity via ligand-dependent transcription. Adeno-associated virus (AAV) vectors enable gene therapy through efficient gene delivery. MicroRNAs (miRNAs) fine-tune immune responses via post-transcriptional silencing. This review summarizes the signaling pathway, cytokine storm, and neuroinflammation induced by the NLRP3 inflammasome, as well as combination therapies with NRs, AAVs, and miRNA.
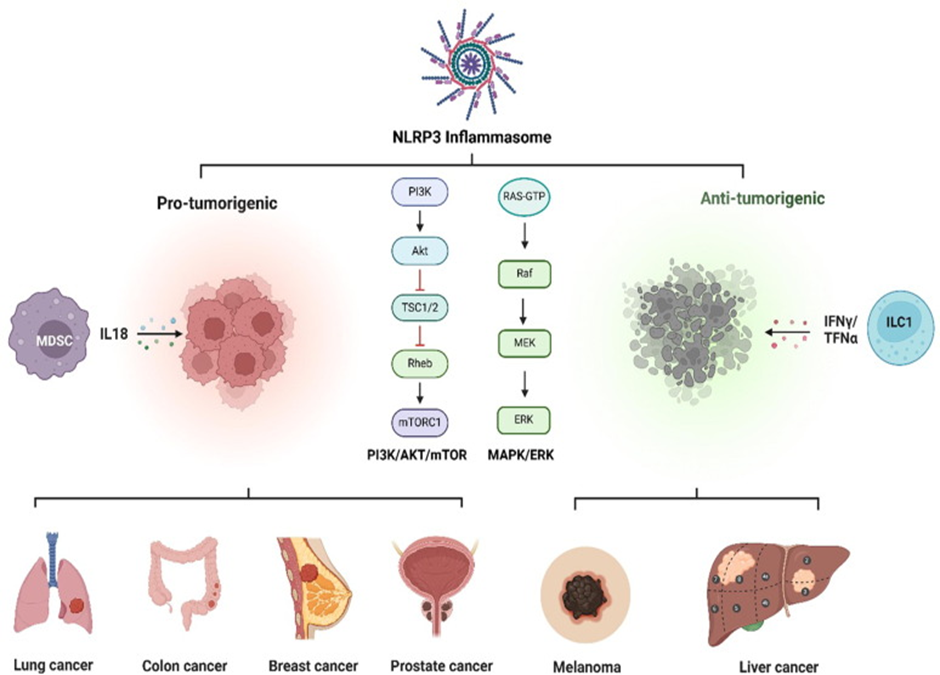
Figure 1. NLRP3-triggered signaling pathways have dual effects in tumor formation and development: both pro-tumorigenic (lung cancer, colon cancer, breast cancer, prostate cancer) and anti-tumorigenic (liver cancer, melanoma).
NLRP3-triggered cytokine storms and COVID-19
Coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in a large global pandemic through respiratory droplets and close contact. COVID-19 is characterized by a diverse range of symptoms, such as fever, chills, cough, dyspnea, fatigue, sore throat, and body pain, as well as multiple organ dysfunction syndrome (MODS, such as respiratory failure, circulatory failure, and liver function injury). Globally, JN.1 is the most reported variant of interest (VOI), and has been found in 139 countries. KP.3.1.1 is showing increasing prevalence, whereas KP.3, KP.2, and JN.1.7 are showing decreasing prevalence
A cytokine storm is an acute overproduction and uncontrolled release of pro-inflammatory cytokines, such as IL-1, IL-2, IL-6, IL-10, TNF-α, IFN-γ, IP-10, MCP-1, and GM-CSF, and the term was first used in 1993 to describe graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. IP-10, Interferon Gamma-Induced Protein 10/CXCL10, is a CXC chemokine secreted by IFN-γ-stimulated monocytes, endothelial cells, and fibroblasts, which binds the CXCR3 receptor to recruit activated T cells, NK cells, and monocytes to sites of inflammation or infection. MCP-1, Monocyte Chemoattractant Protein 1/CCL2, is a CC chemokine produced by macrophages, epithelial cells, and vascular endothelium, which signals via CCR2 to recruit monocytes, memory T cells, and dendritic cells to inflamed or injured tissues. GM-CSF, Granulocyte-Macrophage Colony-Stimulating Factor, is a hematopoietic cytokine secreted by T cells, macrophages, and endothelial cells, which promotes myeloid cell differentiation (granulocytes, monocytes/macrophages) and enhances their phagocytic, antigen-presenting, and pro-inflammatory functions. SARS-CoV-2 cellular entry – which depends on the binding of S proteins covering the virion surface with cellular angiotensin-converting enzyme 2 (ACE2) receptor and on S protein priming by transmembrane protease serine 2 (TMPRSS2) – could trigger immune responses, including a cytokine storm, as well as acute respiratory distress syndrome (ARDS) and death. NLRP3 inflammasome activation during SARS-CoV-2 infection triggers cytokine storm and pyroptosis via caspase-1-mediated mechanisms, including GSDMD cleavage, IL-1β/IL-18 maturation, and DAMP release. Consequently, NLRP3 inflammasome inhibitors could be applied to controlling cytokine storms in patients with SARS-CoV-2 infection.

Figure 2. Activation of the NLRP3 inflammasome by SARS-CoV-2 infection could lead to a cytokine storm. The NLRP3 inhibitor MCC950 could mitigate SARS-CoV-2-mediated inflammatory cytokine signaling by targeting NLRP3.
JAK/STAT Pathway
The JAK/STAT pathway is a key regulator of immune cell function and inflammation. Activation of the JAK/STAT pathway leads to the transcription of genes involved in immune response and inflammation.
JAK/STAT inhibition is revolutionary for autoimmune diseases, providing targeted, oral, and effective alternatives to broad immunosuppressants.
MAPK Pathway
The MAPK pathway is a key regulator of cell signaling and inflammation. Activation of the MAPK pathway leads to the transcription of genes involved in inflammation, immune response, and cell survival.
MAPK (specifically p38 MAPK) pathways are critical in regulating microglia and astrocyte activation in neuroinflammation.
Mitogen-activated protein kinase (MAPK)
It is widely recognized that MAPKs, which include p38 MAPK, ERK, and c-Jun NH2-terminal kinases (JNK), as well as their isoforms ,play a critical role in the regulation of various biological processes, including proliferation, differentiation, apoptosis and inflammation in mammalian cells .The MAPK signaling cascade comprises a MAPKK kinase, a MAPK kinase, and a MAP kinase that respond to both internal and external stimuli, such as growth factors, cytokines, oxidation, and endoplasmic reticulum stress. Activation of the MAPK signaling pathway has been observed in the brains of patients with AD and animal models . In vitro studies have shown that stimulation of Aβ induces the activation of this pathway in glial cell cultures, indicating its involvement in the development of AD Inhibition of tau kinases, such as p38 MAPK, has been shown to improve cognitive deficits and reduce tau pathology in AD Furthermore, blocking the ERK pathway can reverse mitochondrial dysfunction in AD , while specific JNK inhibitors can enhance synaptic function. Of note, the MAPK signaling pathway can also regulate the neuroinflammatory response of microglia. Aβ-induced production of inflammatory cytokines and ROS can activate this pathway, leading to more severe inflammation. A number of in vitro experiments have demonstrated that inhibition of the MAPK signaling pathway can suppress neuroinflammation in BV2 microglia highlighting its potential as an effective strategy for treating AD (Fig. 2 and Table II).
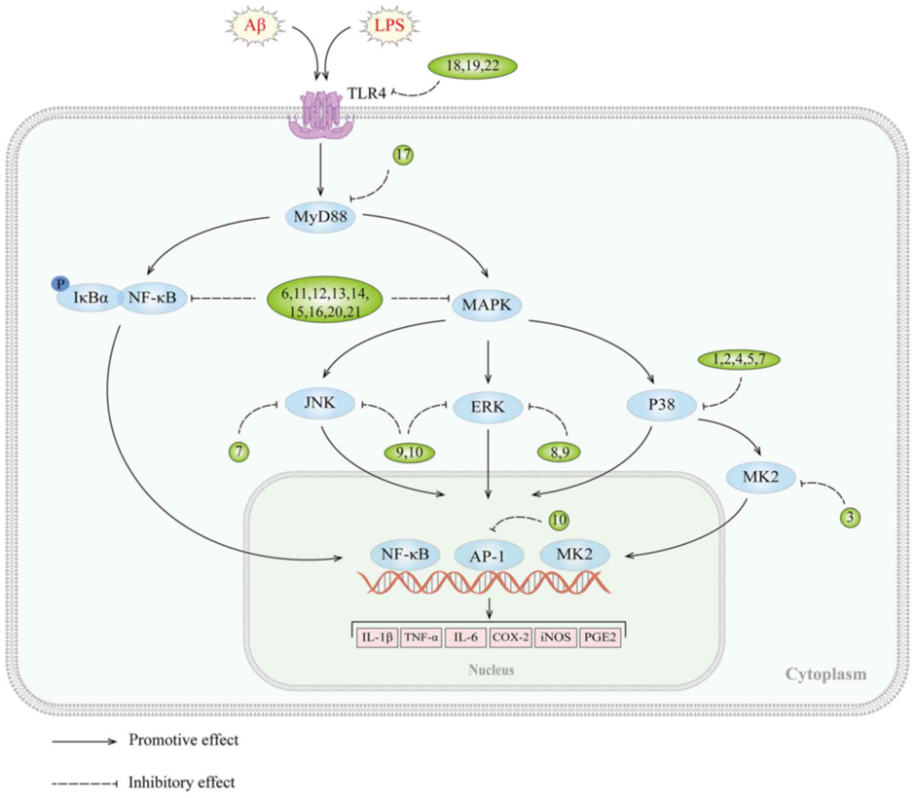
Figure 2
Therapeutic Strategies
Several therapeutic strategies have been developed to target the NF-KB, NLRP3, JAK/STAT, and MAPK pathways, including:
Small Molecule Inhibitors
Small molecule inhibitors have been developed to target specific kinases or enzymes involved in the NF-KB, NLRP3, JAK/STAT, and MAPK pathways. These inhibitors have shown promise in reducing inflammation and improving symptoms in preclinical models.
Gene and RNA-Based Therapies
Gene and RNA-based therapies have been developed to target specific genes involved in inflammation. These therapies include siRNA, CRISPR, and ASOs, which have shown promise in reducing inflammation and improving symptoms in preclinical models.
Stem Cell Therapies
Stem cell therapies have been developed to modulate immune responses and promote repair. Mesenchymal stem cells and induced pluripotent stem cells have shown promise in reducing inflammation and improving symptoms in preclinical models.
Epigenetic Modifications
Epigenetic modifications play a critical role in regulating gene expression and inflammation. Targeting epigenetic modifications, such as histone modifications and DNA methylation, has shown promise in reducing inflammation and improving symptoms in preclinical models.
Table 1:Therapeutic Strategies
|
Sr.no |
Pathways |
Key components |
Function |
|
1. |
NF-KB |
IKK,IKB,NF-KB |
Pro-inflammatory gene expression |
|
2. |
NLRP3 |
NLRP3, ASC, caspase-1 |
Inflammasome activation, pro-inflammatory cytokine cleavage |
|
3. |
JAK/ STAT |
JAK,STAT |
Cytokine signaling, immune response |
|
4. |
MAPK |
MAPKKK, MAPKK, MAPK |
Cell signaling, proliferation, differentiation. |
Summary of Emerging Trends
CONCLUSION
Recent advances in understanding the molecular mechanisms underlying inflammation have led to the development of novel therapeutic strategies targeting key signaling pathways. Further research is needed to translate these findings into clinically viable therapies.
FUTURE DIRECTIONS
Further research is needed to understand the complex interplay between the NF-KB, NLRP3, JAK/STAT, and MAPK pathways and their role in specific diseases.
Development of novel therapeutic strategies targeting these pathways may lead to more effective treatments for chronic inflammatory diseases.
REFERENCES

Pratiksha Patil, S. A. Deshmukh, R. S. Bachhav, Bhakti Pawar, Madhuri Damale, Recent Advances in Molecular Pathways Targeted by Anti-Inflammatory Agents (NF-KB, NLRP3, JAK/STAT, MAPK), Int. J. of Pharm. Sci., 2026, Vol 4, Issue 2, 1-10. https://doi.org/10.5281/zenodo.18452874
 10.5281/zenodo.18452874
10.5281/zenodo.18452874