
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Research Scholar, Guru Nanak College of Pharmaceutical Sciences, Dehradun
2Associate Professor, Guru Nanak College of Pharmaceutical Sciences, Dehradun
Pharmacovigilance (PV) is the science dedicated to the detection, assessment, understanding, and prevention of adverse drug reactions (ADRs) and other drug-related problems throughout a medicinal product’s lifecycle. This comprehensive overview traces the evolution of drug safety from the thalidomide tragedy of the 1960s—which catalyzed foundational regulations such as the Kefauver-Harris Amendments and the WHO Programme for International Drug Monitoring—to modern frameworks including ICH guidelines (E2A, E6(R3), E2F, E8(R1), E19), CIOMS VI, and regional regulations like the EU Clinical Trials Regulation 536/2014 and UK MHRA requirements. The text details safety surveillance across preclinical, Phases I–IV, and post-marketing settings, distinguishing investigator-initiated trials (IITs) from industry-sponsored studies, and outlines advanced safety monitoring practices: signal detection using disproportionality analysis (ROR, IC), the role of Data Safety Monitoring Boards (DSMBs), safety data collection methods and databases (FAERS, Surveillance, VigiBase), and special considerations for pediatric, geriatric, and pregnant populations. Finally, it highlights innovations in electronic systems, artificial intelligence, and real-world data integration that enable near-real-time, globally harmonized pharmacovigilance, ultimately protecting patient safety and informing regulatory decision-making.
Introduction to Pharmacovigilance
Pharmacovigilance, derived from the Greek ‘pharmakon’ (drug) and Latin ‘vigilare’ (to keep watch), is the pharmacological science and set of activities relating to the detection[1], assessment, understanding, and prevention of adverse effects or any other drug-related problem. Its definition has expanded beyond merely monitoring side effects to encompass a holistic, patient-safety-centered approach that spans the entire lifecycle of a medicinal product. The core objectives of PV are fundamentally oriented towards safeguarding public health[2][3]. Firstly, it aims to improve patient care and safety by monitoring the risk-benefit balance of drugs as they are used in real-world populations, which often differ significantly from controlled clinical trial participants. Secondly, PV seeks to identify and quantify previously unrecognized adverse drug reactions (ADRs), particularly serious or fatal ones, and to understand their mechanisms. Thirdly, it is responsible for disseminating actionable safety information to healthcare professionals and patients, thereby preventing unnecessary harm. Finally, an overarching objective is to contribute to the effective regulation of medicines, ensuring that marketing authorizations are maintained, modified, or revoked based on robust safety evidence. In essence, PV transforms passive observation of drug harm into an active, systematic, and scientific discipline that informs clinical practice and regulatory decision-making at every stage[4][5].
The Evolution of Drug Safety (From Thalidomide to Modern Regulations)
The history of modern pharmacovigilance is irrevocably tied to the thalidomide tragedy of the late 1950s and early 1960s. Thalidomide, a sedative and antiemetic widely prescribed to pregnant women for morning sickness, was later found to cause severe phocomelia (limb malformations) in thousands of newborns across 46 countries. This global catastrophe exposed the catastrophic inadequacy of then-prevailing drug safety frameworks[6][7], which relied almost exclusively on rudimentary animal toxicology and spontaneous case reports without mandatory pre-marketing clinical trials or post-marketing surveillance. The disaster catalyzed a paradigm shift. In the United States, Dr. Frances Kelsey’s refusal to approve thalidomide due to insufficient safety data led directly to the Kefauver-Harris Amendments of 1962, which for the first time required drug manufacturers to prove both safety and efficacy through “substantial evidence” from adequate and well-controlled clinical trials before marketing. Internationally, the World Health Organization (WHO) launched its Pilot Research Project for International Drug Monitoring in 1968, evolving into the WHO Programme for International Drug Monitoring, which now coordinates VigiBase, a global database of individual case safety reports. Subsequent decades saw further evolution driven by other crises, such as the recognition of serious cardiovascular risks with non-steroidal anti-inflammatory drugs (e.g., rofecoxib withdrawal in 2004), leading to the establishment of proactive risk management plans (RMPs), pregnancy registries, and advanced signal detection methodologies. Today, regulations like the EU’s Good Pharmacovigilance Practices (GVP) modules and FDA’s Sentinel Initiative represent the culmination of this evolution, mandating continuous, lifecycle safety monitoring using real-world data and sophisticated epidemiological methods[8].
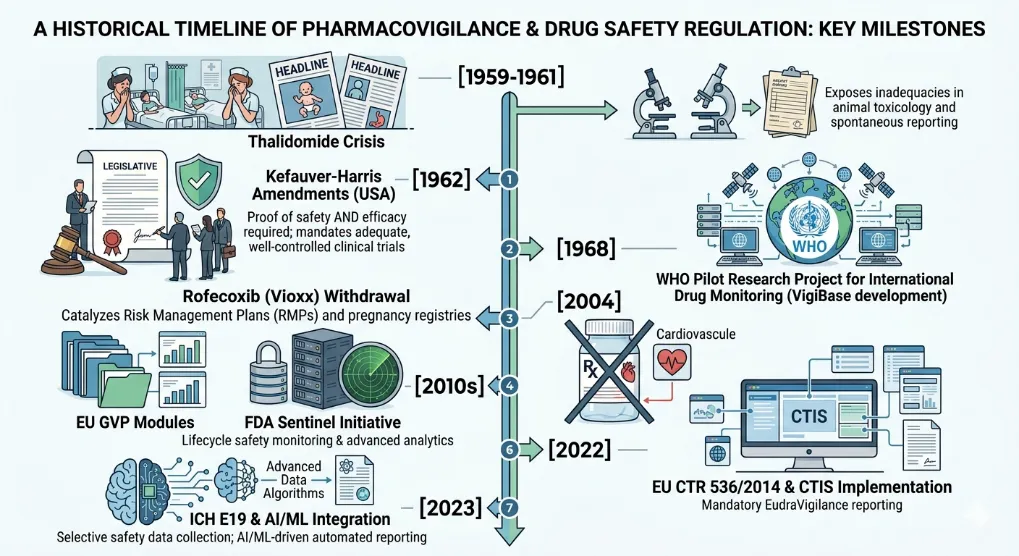
Fig: 1 Evolution of Pharmacovigilance & Drug Safety
Key Regulatory Frameworks (ICH, FDA, EMA, WHO)
The global practice of pharmacovigilance is harmonized and enforced by several key regulatory frameworks. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) is the most critical body, bringing together regulatory authorities and pharmaceutical industry representatives from Europe, Japan, and the United States. Its guidelines, particularly the E2 series on pharmacovigilance, are the global gold standard. Key ICH guidelines include E2A on clinical safety data management (defining expedited reporting timelines for serious, unexpected ADRs), E2B on electronic transmission of individual case safety reports, E2C on periodic benefit-risk evaluation reports (PBRERs), and E2E on pharmacovigilance planning[6]. The U.S. Food and Drug Administration (FDA) enforces its framework through the Code of Federal Regulations (21 CFR), utilizing systems like FAERS (FDA Adverse Event Reporting System) and Sentinel, and mandates Risk Evaluation and Mitigation Strategies (REMS) for high-risk drugs[9][10]. The European Medicines Agency (EMA) operates a centralized, network-based system with the EU’s national competent authorities, governed by the comprehensive Good Pharmacovigilance Practices (GVP) modules, which are arguably the most detailed PV guidelines worldwide. The EMA manages EudraVigilance, the European database of suspected ADRs, and the PRAC (Pharmacovigilance Risk Assessment Committee) for independent safety assessments. The World Health Organization (WHO), while not a regulatory authority, provides a crucial coordinating role, especially for low- and middle-income countries, through its Uppsala Monitoring Centre (UMC) which manages VigiBase and promotes standardized MedDRA (Medical Dictionary for Regulatory Activities) coding. These frameworks collectively ensure that wherever a drug is developed or used, the core principles of timely reporting, rigorous signal detection, and transparent risk communication are applied, despite local legal variations[11][12].
Ethical & Good Clinical Practice (GCP) Principles
The ethical foundation of pharmacovigilance is inseparable from the principles of Good Clinical Practice (GCP), as codified by the ICH E6(R2) guideline, and the Declaration of Helsinki. The paramount ethical principle is patient safety and the protection of human subjects. In the context of PV, this translates into a non-negotiable duty to promptly report any serious, unexpected adverse event, regardless of suspected causality, to regulatory authorities and ethics committees[13][14]. The second principle is informed consent; participants in clinical trials must be explicitly informed about the nature of adverse events, the procedures for monitoring their safety, and the possibility of receiving information about significant new risks that emerge during the trial. Thirdly, the principle of beneficence (acting in the patient’s best interest) requires that the risk-benefit ratio be continuously reassessed throughout a drug’s lifecycle. If new safety signals emerge that tip this balance negatively, ethical obligations may require protocol modification, halting a trial, or withdrawing a marketed drug[15][16]. Fourth, justice and equity demand that vulnerable populations (e.g., pregnant women, children, the elderly, and those with comorbidities) are not systematically excluded from safety surveillance but rather are actively monitored. GCP principles also mandate rigorous data integrity and confidentiality; adverse event reports must be accurate, verifiable, and managed with respect for patient privacy. The ethics of PV extend to publication and transparency; negative safety findings must be disclosed alongside positive efficacy results to prevent outcome reporting bias. Ultimately, GCP provides the operational framework—including standard operating procedures for safety reporting, quality control, and audit trails—that transforms abstract ethical commitments into daily, verifiable practices in clinical research and post-marketing surveillance[17][18].
Extending Safety Surveillance from Preclinical to Post-Marketing
Safety surveillance is not a discrete phase but a continuous thread that connects preclinical research to post-marketing monitoring. In the preclinical stage, safety assessment begins with in vitro toxicology studies (e.g., hERG channel assays to predict cardiac arrhythmia risk) and in vivo animal studies (e.g., repeated-dose toxicity, reproductive toxicology, carcinogenicity).
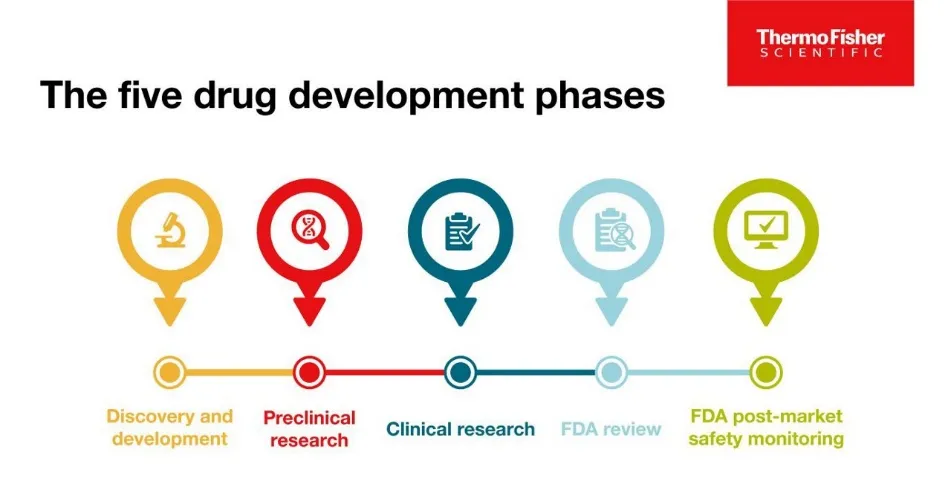
Fig: 2 Stage of drug discovery
These studies identify target organs of toxicity, establish dose-limiting toxicities (e.g., NOAEL – No Observed Adverse Effect Level), and help define a safe starting dose for first-in-human trials. However, animal models have well-recognized limitations, failing to predict rare idiosyncratic reactions (e.g., Stevens-Johnson syndrome)[19] or toxicities that are uniquely human. As a drug moves into clinical trials (Phases I-III), safety surveillance intensifies with structured data collection, adjudication of serious adverse events, and predefined stopping rules. Yet, even large Phase III trials (typically 3,000-5,000 patients) are statistically underpowered to detect ADRs with an incidence of less than 1 in 1,000, and they exclude vulnerable populations, short-term treatment periods, and lack polypharmacy interactions. This is why safety surveillance must robustly extend into the post-marketing (Phase IV) period. Here, real-world data from spontaneous reporting systems (e.g., FAERS, EudraVigilance), electronic health records, registries, and observational studies enable detection of rare ADRs (e.g., 1 in 10,000), long-term toxicities (e.g., drug-induced cancers or valvular heart disease), and safety signals in special populations like pregnant women, children, and the elderly[20]. The lifecycle approach mandates that new safety findings from Phase IV trigger re-evaluation of preclinical and clinical data (e.g., mechanistic toxicology studies), leading to label changes, restricted use, or withdrawal. This bidirectional flow of information—from bench to bedside and back—is the essence of modern, integrated safety surveillance.
The Drug Development Lifecycle (Phases I-IV of Clinical Trials)
The drug development lifecycle is a structured, sequential process designed to progressively characterize a drug’s safety and efficacy, with pharmacovigilance activities embedded in each phase. Phase I trials are the first human exposure, typically involving 20-100 healthy volunteers (or patients with advanced cancer for cytotoxic drugs). The primary PV objectives here are to identify the maximum tolerated dose (MTD), dose-limiting toxicities (DLTs), and the acute safety profile[21][22]. Frequent blood sampling, continuous ECG monitoring, and intensive adverse event (AE) collection characterize this phase. Any serious, unexpected AE is immediately reported to regulators. Phase II trials expand to 100-500 patients with the target disease, aiming to establish preliminary efficacy and short-term safety at therapeutic doses. Pharmacovigilance here focuses on identifying common ADRs (incidence >1-10%), characterizing dose-response for safety, and refining the protocol’s stopping rules. Phase III is the pivotal, confirmatory phase involving hundreds to thousands of patients (often 1,000-5,000) in randomized, controlled trials (RCTs). This is the primary source for pre-marketing safety data, aiming to detect relatively common ADRs, characterize serious events, and generate the safety database that forms the basis of the product label (SmPC or USPI). PV activities include rigorous adjudication of serious events, formal interim analyses, and preparation of the Integrated Summary of Safety (ISS). Phase IV begins after marketing authorization. This post-marketing surveillance phase is the largest and longest, with potentially millions of patients exposed. Key PV tools in Phase IV include spontaneous reporting systems (to generate hypotheses and detect rare ADRs), active surveillance (e.g., registries, pregnancy exposure registries), and large, pragmatic clinical trials or observational studies (e.g., case-control or cohort studies) to quantify risk. Phase IV also mandates periodic safety update reports (PSURs/PBRERs) and the implementation of Risk Management Plans (RMPs). A critical concept is that safety evaluation never truly ends; even in Phase IV, persistent uncertainty may trigger mandated post-marketing requirement studies (PMRs) or, in extreme cases, withdrawal of the drug[23][24].
Investigator-Initiated Trials (IITs) vs. Industry-Sponsored Trials
A crucial distinction in clinical research, with significant pharmacovigilance implications, lies between Investigator-Initiated Trials (IITs) and Industry-Sponsored Trials. In a traditional industry-sponsored trial, the pharmaceutical or biotechnology company holds the Investigational New Drug (IND) application (in the US) or Clinical Trial Authorization (CTA) (in the EU). The sponsor retains ultimate legal and financial responsibility for all pharmacovigilance activities: ensuring proper AE case definition, severity and causality assessment, expedited reporting to regulators and ethics committees, data quality, and safety signal management. The company's global safety database is the central repository, and the sponsor’s Qualified Person for Pharmacovigilance (QPPV) in Europe (or similar role in the US) is legally accountable. The investigator’s role is to comply with the protocol, report AEs promptly to the sponsor, and cooperate with audits[25][26]. Conversely, in an IIT, the investigator (typically an academic physician or research institution) acts as both the investigator and the sponsor. This means the academic sponsor assumes all pharmacovigilance responsibilities, including setting up a safety management system, maintaining an IND/CTA, purchasing insurance, establishing AE reporting workflows, ensuring regulatory compliance (e.g., 15-day expedited reporting for serious, unexpected AEs), and managing signal detection. This distinction creates significant practical challenges. IITs often lack the dedicated pharmacovigilance infrastructure, software, and trained personnel (e.g., drug safety associates, medical reviewers) that industry sponsors routinely maintain. Consequently, IITs are at higher risk for underreporting of AEs, delayed reporting, and non-compliance with global regulations. To mitigate this, many IITs now enter into formal agreements with pharmaceutical companies (e.g., for investigator-initiated studies of an approved drug) where the company provides the investigational product but the academic sponsor retains PV responsibility under a detailed safety management plan. In other cases, contract research organizations (CROs) with PV expertise are hired. Regardless, regulators treat IITs with equal seriousness; the same ICH GCP and PV guidelines apply, but the burden of expertise, resources, and legal liability falls on the academic investigator—a fact that is often underestimated when planning such trials[27][28]. Therefore, robust pharmacovigilance planning is not an optional add-on for IITs but a core, resourced, and auditable component that distinguishes compliant, ethical research from high-risk, potentially harmful non-compliance.
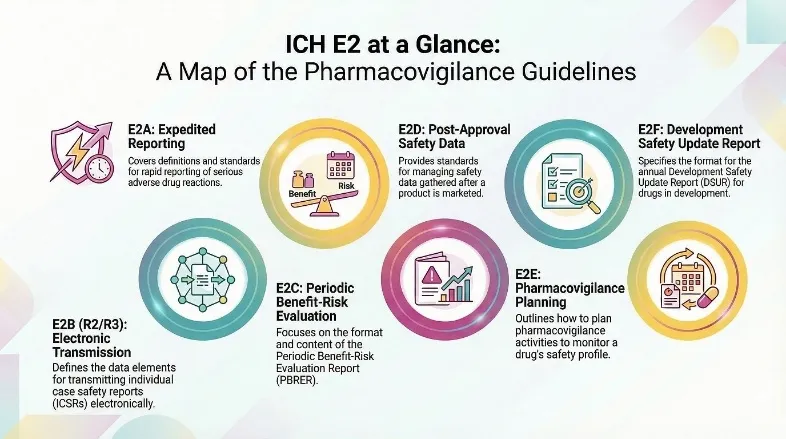
Fig: 3 ICH Guidelines and Their Roles in Pharmacovigilance
Regulatory & Guideline Overview
The regulatory landscape governing clinical trial safety and conduct is built upon a foundation of interconnected ICH guidelines and regional regulations, each designed to protect trial participants, ensure data integrity, and harmonize global standards. Central to this framework is ICH E2A: Clinical Safety Data Management (Definitions & Expedited Reporting) , which establishes critical definitions for adverse events (AEs), adverse reactions (ADRs), and unexpected ADRs, and sets uniform procedures for rapid safety reporting during clinical development. The guideline mandates that all serious, unexpected adverse drug reactions (SUSARs) must be reported to regulators and investigators on an expedited basis—within 7 days for fatal or life-threatening events and 15 days for all other serious reactions—to ensure immediate awareness of new safety signals[29]. Complementing E2A’s focus on safety data, ICH E6(R3): Good Clinical Practice (GCP) has recently been updated to shift sponsor responsibilities from reactive issue correction to proactive, risk-based oversight across the entire trial lifecycle[30][31]. The revised guideline emphasizes “Quality by Design” (QbD), requiring sponsors to identify Critical-to-Quality (CtQ) factors early, implement risk-based quality management (RBQM), and maintain end-to-end accountability even when tasks are outsourced. This proactive framework ensures the rights, safety, and well-being of participants while enhancing data reliability through centralized monitoring and traceable decision-making[32]. Integrating with these principles, ICH E2F: Development Safety Update Report (DSUR) provides a globally harmonized format for annual cumulative safety report ing, replacing disparate regional annual reports such as the US IND Annual Report and the EU Annual Safety Report. The DSUR is submitted annually by sponsors to regulatory authorities, presenting a cumulative analysis of all relevant safety data gathered during the reporting period, including serious adverse events, exposure data, emerging safety signals, and a benefit-risk assessment[33][34]. It also serves to update the Investigator’s Brochure (IB) and reference safety information, ensuring continuous monitoring and risk mitigation throughout the product lifecycle[35]. At a strategic level, ICH E8(R1): General Considerations for Clinical Trials serves as an overarching framework that integrates all the other guidelines[36]. Updated in 2021 after 24 years, E8(R1) introduces the concept of “quality by design,” emphasizing the identification of critical quality factors and proactive risk management during study planning, while also promoting diversity in trial designs, data sources, and stakeholder engagement. Its aim is to ensure participant protection, data integrity, and regulatory acceptance of results without imposing unnecessary complexity or burden. Further optimizing trial efficiency is ICH E19: Selective Approach to Safety Data Collection, a 2023 guideline that permits a risk-proportionate reduction in safety data collection for late-stage pre-approval or post-approval studies where a drug’s safety profile is already well-characterized. Under ICH E19, sponsors may collect only essential safety information—such as serious adverse events, discontinuation-related events, and events of special interest—while omitting routine, non-critical data collection to improve trial feasibility and enrollment[36][37]. These ICH principles are reinforced by the recommendations of CIOMS VI: Management of Safety Information from Clinical Trials, which shifted the focus from post-marketing spontaneous reporting to systematic management of safety information from the earliest clinical trials onward. CIOMS VI emphasizes a holistic approach encompassing ethical oversight, data collection, evaluation, statistical analysis, regulatory reporting, and risk communication, and notably recommended the unified DSUR format that was later codified by ICH E2F. Finally, this ICH-CIOMS framework is implemented regionally, most notably through the EU Clinical Trials Regulation (CTR) 536/2014, which fully replaced the Clinical Trials Directive in January 2022. The CTR mandates direct reporting of all SUSARs to the EudraVigilance database (EVCTM), harmonized annual safety reporting via the DSUR, and submission of both through the centralized Clinical Trial Information System (CTIS)[38][39]. In the United Kingdom, following Brexit, post-transition requirements largely mirror EU standards, with the MHRA expecting sponsors to continue using DSURs and ICH-aligned expedited reporting to ensure participant safety. Collectively, these guidelines and regulations create a cohesive global system that prioritizes proactive risk management, data quality, and patient protection across the entire drug development continuum[40].
Advanced Safety Monitoring
The systematic identification, evaluation, and management of safety signals forms the cornerstone of pharmacovigilance, beginning with Signal Detection & Management—a dynamic process that transforms raw adverse event data into actionable risk knowledge. Signal detection relies primarily on quantitative disproportionality analysis from large spontaneous reporting databases, where observed-to-expected ratios such as the Reporting Odds Ratio (ROR) or Information Component (IC) flag potential associations between a drug and an adverse event[41]. However, statistical alerts alone do not constitute signals; each potential signal must undergo rigorous triage, validation, and prioritization based on clinical relevance, seriousness, strength of association, biological plausibility, and frequency relative to background rates. For example, a robust signal of drug-induced liver injury would require not merely a statistical excess but also consistent temporality, dose-response evidence, and supportive literature before progressing to confirmation and risk mitigation. Modern signal management follows a structured workflow—detection, validation, prioritization, evaluation, and recommendation—integrating data from clinical trials, spontaneous reports, electronic health records, and real-world evidence, with the ultimate goal of updating product labeling, implementing risk minimization measures, or, in rare cases, withdrawal from the market. Parallel to signal detection in clinical trial settings is The Role of Data Safety Monitoring Boards (DSMBs) , independent groups of experts (clinicians, biostatisticians, ethicists) appointed by sponsors to provide real-time oversight of ongoing randomized controlled trials, especially those involving high-risk populations or interventions with uncertain safety profiles. DSMBs operate under a pre-specified charter, gaining access to unblinded interim data at defined intervals—such as after a certain percentage of primary outcomes or at predetermined calendar times—to evaluate both efficacy and safety endpoints[42]. Their core responsibilities include recommending continuation, modification, or termination of a trial based on evidence of overwhelming benefit, unacceptable harm (e.g., significantly increased mortality or serious adverse events), or futility (unlikely to achieve its objectives). Crucially, DSMBs ensure ethical conduct by protecting participants from extended exposure to a harmful intervention or unnecessary placebo while preserving trial integrity through strict confidentiality and operational independence from the sponsor. For example, in a cardiovascular outcomes trial of a new antidiabetic agent, the DSMB might receive unblinded comparisons of major adverse cardiac events; if an unacceptably large imbalance emerges, they can recommend early trial termination and unblinding for patient safety. The infrastructure enabling such oversight relies on robust Safety Data Collection Methods and Databases, which have evolved from simple paper case report forms to sophisticated electronic data capture systems integrated with medical coding dictionaries like MedDRA (for adverse events) and WHO Drug Global (for medicinal products). In clinical trials, safety data collection typically begins at screening and continues through treatment and follow-up, capturing adverse events (AEs), serious adverse events (SAEs), laboratory abnormalities, vital signs, physical examination findings, and concomitant medications. Sources include investigator-reported observations, patient diaries, laboratory databases, and increasingly, direct patient-reported outcomes via ePRO (electronic patient-reported outcomes) portals[43]. For post-marketing surveillance, spontaneous reporting remains the primary method, where healthcare professionals or patients submit individual case safety reports (ICSRs) to national pharmacovigilance centers or marketing authorization holders. However, passive reporting is supplemented by active surveillance methods, including targeted clinical investigations, prescription-event monitoring (PEM), and linkage of electronic health records (EHRs) or claims databases. Safety databases—such as the FDA Adverse Event Reporting System (FAERS) or EudraVigilance—aggregate millions of ICSRs, enabling continuous statistical monitoring. These databases employ structured data elements (e.g., patient age, sex, drug exposure dates, outcome) and follow the ICH E2B(R3) format for electronic transmission of ICSRs, ensuring global interoperability and standardized case processing[44]. A critical dimension of pharmacovigilance is the assessment of Pharmacovigilance in Specific Populations (Pediatrics, Geriatrics, Pregnancy) , each presenting unique physiological, pharmacokinetic, and ethical challenges. In pediatrics, children are not “small adults”; their developing organ systems (especially hepatic and renal function) and age-dependent metabolism require age-specific dosing and safety monitoring. Adverse drug reactions (ADRs) in neonates may differ dramatically from those in adolescents; for example, valproate-induced liver toxicity manifests more severely in young children, while growth suppression from corticosteroids is a pediatric-specific concern. Regulatory frameworks, including the Pediatric Research Equity Act (PREA) and EU Pediatric Regulation, mandate pediatric drug development and dedicated safety studies, with dosing extrapolation only when disease progression and drug response are sufficiently similar to adults. In geriatrics, polypharmacy, age-related decline in renal and hepatic clearance, sarcopenia, and increased susceptibility to falls, delirium, and orthostatic hypotension demand proactive safety assessment. Older adults are often excluded from clinical trials, leading to post-marketing safety signals that may go undetected; therefore, geriatric-specific pharmacovigilance emphasizes anticholinergic burden, sedative-hypnotic risks (e.g., benzodiazepines), and drug-drug interactions with anticoagulants or antihypertensives. For pregnancy and lactation, the safety of medications poses a dual challenge—protecting the fetus/neonate from teratogenicity or neonatal withdrawal syndromes while managing maternal health conditions. Regulatory agencies maintain pregnancy exposure registries and require enhanced pharmacovigilance through labeling (e.g., FDA’s Pregnancy and Lactation Labeling Rule, which replaces letter categories with more descriptive risk summaries). Signal detection for pregnancy outcomes requires careful differentiation of drug effects from underlying disease, as well as handling of outcomes such as spontaneous abortion, preterm birth, low birth weight, and major congenital malformations. Modern approaches use linked data from birth registries, claims databases, and EHRs to support high-quality pharmacoepidemiologic studies, but spontaneous reporting still provides early alerts (e.g., the link between isotretinoin and severe birth defects leading to risk management programs like iPLEDGE)[45]. Underpinning all these activities are Electronic Systems and Innovations (e.g., EudraVigilance, VigiBase) , which have transformed pharmacovigilance from manual, retrospective case review into near-real-time, globally integrated surveillance. EudraVigilance is the European Medicines Agency’s system for managing and analyzing suspected adverse reaction reports to authorized medicinal products in the European Economic Area. It consists of two complementary modules: EV post-authorisation module (for marketing authorization holders to submit ICSRs) and EV Clinical Trial Module (EVCTM), which under the EU Clinical Trials Regulation 536/2014 became the mandatory single entry point for all SUSARs from clinical trials in the EU[46][47]. EudraVigilance employs advanced data analytics, including the proportional reporting ratio (PRR) and Bayesian confidence propagation neural network (BCPNN) for signal detection, and provides public access via the EudraVigilance Expert Working Group’s public dashboards. Crucially, it enforces de-duplication and data quality by requiring R3 format submission and automatically linking multiple reports from the same patient or same reaction across different products[48]. In parallel, VigiBase, maintained by the Uppsala Monitoring Centre (UMC) on behalf of the WHO Programme for International Drug Monitoring, is the largest global database of ICSRs, containing over 30 million reports from more than 170 member countries. Unlike EudraVigilance’s focus on the EU, VigiBase covers all regions, enabling detection of rare signals, cross-cultural patterns of drug use, and safety issues in settings where clinical trials may not be conducted. VigiBase employs the standardized WHO Drug Dictionary and MedDRA, and it uses the BCPNN to generate disproportionality scores (Information Components) that evolve as new data accumulate[49]. Beyond these two core systems, innovations include the use of artificial intelligence (AI) for automated narrative extraction from clinical notes, natural language processing to map unstructured text to MedDRA terms, machine learning for duplicate detection and causality assessment, and blockchain for traceable, tamper-proof safety data exchange. Electronic medical records are increasingly integrated with pharmacovigilance systems through common data models (e.g., Observational Medical Outcomes Partnership, OMOP), enabling near-real-time active surveillance[50]. Collectively, these digital ecosystems enable regulators and sponsors to detect signals earlier, prioritize higher-risk drugs, and ultimately protect populations across all ages and conditions—from neonates to older adults, and from early-phase trials to decades of real-world use.
CONCLUSION
Modern pharmacovigilance has matured from a passive, post-marketing activity into an integrated, proactive, and lifecycle-wide scientific discipline. The foundational lessons of the thalidomide disaster permanently embedded the principle that drug safety cannot be assured by pre-marketing clinical trials alone; instead, continuous surveillance across preclinical, clinical, and real-world phases is mandatory. Key ICH guidelines (E2A, E6(R3), E2F, E8(R1), E19) and CIOMS VI provide a harmonized global framework for expedited reporting, risk-based quality management, cumulative safety reporting (DSUR), and selective data collection. Regional implementation through the EU CTR 536/2014 and UK MHRA requirements further reinforces these standards. Advanced safety monitoring encompassing quantitative signal detection (ROR, IC, BCPNN), independent DSMB oversight, robust safety databases (FAERS, EudraVigilance, VigiBase), and specialized vigilance for pediatrics, geriatrics, and pregnancy—ensures that emerging risks are identified and mitigated promptly. Innovations such as artificial intelligence, natural language processing, and electronic health record integration are transforming PV into a near-real-time, globally interconnected surveillance system. Nevertheless, challenges persist, particularly in investigator-initiated trials where pharmacovigilance infrastructure is often lacking. Ultimately, the core objectives of PV improving patient care, identifying unrecognized ADRs, disseminating actionable safety information, and supporting evidence-based regulation—remain as vital today as ever. A strong ethical foundation rooted in Good Clinical Practice, informed consent, beneficence, and justice ensures that patient safety is never subordinated to other interests. As drug development grows increasingly complex (e.g., cell and gene therapies, personalized medicine), pharmacovigilance must continue to evolve, embracing real-world evidence, digital health tools, and global collaboration to fulfill its enduring mission: keeping watch over medicines to protect human health.
REFERENCES

Vinay Sharma*, Archana Rautela, Artifical Intelligence in Drug Approval and Clinical Drugs, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7439-7451. https://doi.org/10.5281/zenodo.20415218
 10.5281/zenodo.20415218
10.5281/zenodo.20415218