We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, College of Pharmaceutical Sciences,Government Medical College, Thiruvanthapuram.
Benzimidazole, also referred to as 1H-benzimidazole or 1,3-benzodiazole, is made up of a benzene ring that is fused with a five-membered imidazole ring and is a significant heterocyclic pharmacophore. This compound is considered a “privileged structure” in heterocyclic chemistry due to its association with various biological activities. They demonstrate considerable effectiveness against various viruses, encompassing antimicrobial, anti-inflammatory, potential antitumor, antiparasitic, and antiprotozoal agents, as well as HIV, herpes (HSV-1), RNA viruses, influenza, and human cytomegalovirus (HCMV). Recent studies have concentrated on creating new derivatives using both traditional and environmentally friendly chemistry methods, examining their structure–activity relationships (SAR), and assessing their prospects as candidate molecules in drug development. This review summarises the chemical diversity, biological activity, a dual targeting action of benzimidazole analogues and their future perspectives
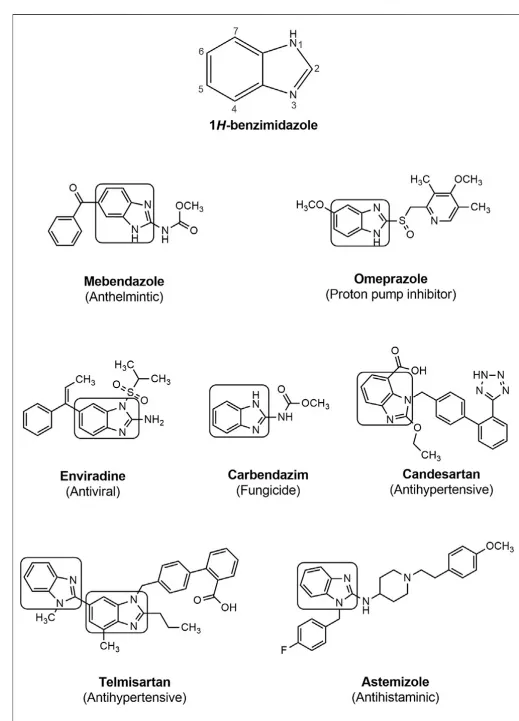
Benzimidazole, alternatively known as 1H-benzimidazole and 1,3-benzodiazole, consists of a benzene ring fused with a five-membered imidazole ring and is an important heterocyclic pharmacophore. Benzimidazole is regarded as a “privileged structure” in heterocyclic chemistry due to its association with a wide range of biological activities 1. They exhibit significant activity against several viruses, including antimicrobial, anti-inflammatory, potential antitumour, antiparasitic, antiprotozoal agents, HIV, herpes (HSV-1), RNA, influenza, and human cytomegalovirus (HCMV) 2. As a result of changing substituents around the core structure, many drugs of a wide variety of therapeutic lines have been developed, such as albendazole, mebendazole, thiabendazole as anthelmintics; enviradene as antiviral; carbendazim as fungicidal; omeprazole, lansoprazole, pantoprazole as proton pump inhibitors; candesartan and telmisartan as antihypertensives, and astemizole as an antihistaminic agent (Figure 1) 3. The diverse biological activities displayed by compounds bearing a benzimidazole moiety have prompted researchers all around the globe to design and synthesize various benzimidazole analogues.
2. Chemistry of the Benzimidazole Scaffold
Benzimidazoles are commonly synthesized by reacting 1,2-diaminobenzene derivatives with carboxylic acids under strongly dehydrating conditions. Traditional methods, such as the Philips method, employ strong acids, including hydrochloric acid, polyphosphoric acid, boric acid, or p-toluene sulfonic acid. In recent years, the use of milder catalysts like Lewis acids, mineral acids, and inorganic clays has improved the reaction by enhancing both product yield and purity. Another important approach involves the condensation of 1,2-diaminobenzenes with aldehydes, which requires an oxidizing agent for cyclization and formation of the benzimidazole ring. Different oxidizing reagents such as nitrobenzene, benzoquinone, sodium metabisulfite, mercuric oxide, lead tetraacetate, iodine, copper(II) acetate, indium perfluoro octane sulfonates, ytterbium perfluoro octane sulfonates, and even atmospheric oxygen have been used successfully for this transformation. Additionally, benzimidazole derivatives can also be synthesized through the coupling of 1,2-diaminobenzenes with carboxylic acid derivatives, including nitriles, imidates, ortho esters, anhydrides, and lactones 4,5.
3. Anticancer Mechanisms of Benzimidazole Derivatives
Cancer is one of the leading health hazards that is affecting a wide majority of the world's population. Various anticancer agents reported for the treatment of various kinds of cancers act through different mechanisms 6. Mahadevi Vitthal Kendre et al. (2026) investigated benzimidazole-based thioamide derivatives and evaluated their anticancer potential, supported by EGFR-targeted molecular docking studies. The synthesis involves SN2 reaction between1-(substitutedphenyl)-2-((5-(difluoromethoxy)-1Hbenzo[d]imidazol-2-yl) thio) ethan-1-one intermediate and brominated nitro benzyl derivative 7. The synthesized compounds were evaluated for in vitro cytotoxic activity against three human cancer cell lines: MCF7 (breast), A-549 (lung), and HEP-G2 (liver). Molecular docking studies in the EGFR kinase domain (PDB ID: 2JIV) revealed favourable binding interactions, superior docking scores when compared against the standard drug Neratinib. The synthesized compounds were evaluated for in vitro cytotoxic activity against three human cancer cell lines: MCF7 (breast), A-549 (lung), and HEP-G2 (liver). After optimizing the solvent (acetonitrile) and base (potassium carbonate) effect on reaction temperature on yield and efficiency of intermediates where calculated and found to be 820C for 6 hours. Among these derivative compounds 1 shows the most potent, displaying IC₅₀ values of 3.70 μm against A-549 and 5.38 μm against HEP-G2 cells.
Compound 1
Boye Jiang et al. (2026) investigated quinazolinone and benzimidazole analogues as tubulin polymerization inhibitors with potent anticancer activities. Tubulin activity was designed via the molecular hybridization technique, and Antiproliferative activities against MCF-7, MDA-MB-231, A549, and HeLa cell lines were evaluated using the CCK8 assay. Apoptosis induction and cell cycle arrest were analysed by flow cytometry. In vivo activity validated in a mouse melanoma tumour model. Compound 2 was the most promising candidate and displayed strong broad-spectrum anticancer activity with an average IC50 value of 2 μm against the MCF-7, MDA-MB-231, A549, and HeLa cell lines 8. Mechanistic studies revealed that compound 3 inhibited tubulin polymerization, arrested the cell cycle at the G2/M phase, and induced apoptosis in MCF-7 cells. Molecular docking analysis indicated that the imidazole ring of compound 3 established an important hydrogen bond interaction with the βLys254 residue of tubulin, while the remaining portion of the molecule fit effectively within the hydrophobic binding pocket formed by surrounding tubulin amino acid residues, suggesting strong binding affinity. These interactions are likely responsible for the potent antitumor activity exhibited by compound 3.
Compound 2
Aladdin. M. Srour et al. (2025) investigated benzimidazole-triazole glycoconjugates as anticancer agents and EGFR inhibitors. The derivatives have been assessed for cytotoxic activity on diverse human cancer cell lines, namely HepG-2 (human liver cancer), HCT-116 (human colorectal), and MCF-7 (human breast cancer), in addition to a human normal cell line (BJ-1), following the LDH assay and with erlotinib and doxorubicin as the standard references. Compound 3 shows the best activity against all the targeted cell lines, HepG-2 and HCT-116 (IC50 1.64±0.11 and 5.00±0.51 µM), greater than standard erlotinib (IC50 =2.07±0.07 and 5.14±0.33µM), and showed a safe profile against the tested normal cell line BJ-1. Compound 3 and compound 5 inhibitory activity against EGFR (IC50 =0.069±0.003 and 0.075±0.003µM, respectively) in comparison with erlotinib, the reference drug (0.048±0.002 µM) 9. Docking studies suggest the incorporation of α, β-unsaturated ketone function enhances compounds’ stability within the EGFR active site.
Compound 3
9b- R1= H, R2 =OH, R3= CH2OH
Seref Demirayak et al. (2010) investigated 2-arylbenzimidazole and pyrazino-benzimidazole derivatives, and in vitro studies were conducted to evaluate IC50 levels in various cell lines. A compound 4(a) bearing a methoxy group shows remarkable anticancer activity 10. The growth inhibitory effects of the compounds were evaluated against approximately sixty human tumour cell lines derived from nine neoplastic diseases namely; Leukemia (L, 4 or 6 cell lines), Non-Small Cell Lung Cancer (NSCLC, 9 cell lines), Colon Cancer (CC, 7 cell lines), Central Nervous System Cancer (CNSC, 6 cell lines), Melanoma (M, 8 or 9 cell lines), Ovarian Cancer (OC, 6 or 7 cell lines), Renal Cancer (RC, 8 cell lines), Prostate Cancer (PC, 2 cell lines), Breast Cancer (BC, 6 or 8 cell lines). A 48-h continuous drug exposure protocol was followed, and a sulforhodamine B (SRB) protein assay was used to estimate cell viability of growth 11,12,13,14.
Compound 4(a, b)
a-R=H, R'=OCH 3;
b-R=OCH 3, R'=OCH3
4. Anti-Inflammatory Mechanisms of Benzimidazole Derivatives
The inhibition of prostaglandin synthesis by blocking the COX pathway is the common basis by which several NSAIDs show therapeutic effects. The COX-2 pathway inhibitors have shown successful reduction in gastrointestinal irritation or haemorrhage compared to the other NSAIDs. Also, they possess an increased risk of myocardial infarction and cardiovascular thrombotic events. The 5-LOX and 15-LOX show undesirable activity in humans, leading to inflammation, hypersensitivity, asthma, and atherosclerosis 15. Compared with simple COX inhibitors, compounds with dual inhibitory activities against COX and 5-LOX, such as KME-4, E-5110, S-2474, Tenidap, CI-987, and 3,5-di-tert-butyl-4-hydroxyphe-nyl derivatives, have been reported to be superior and have been studied as potential anti-inflammatory agents with an improved safety profile 16.Harshali Chonde et al. (2026) investigated 2 phenyl benzimidazole amides through molecular hybridization. From spectroscopic studies and in vivo carrageenan-induced paw edema model studies, compounds 5a, 5b, and 5c show potent anti-inflammatory activity with inhibition values of 95.65, 96.89, and 97.61 % 17. Molecular docking studies were performed in p38 kinase, and simulation studies validated the stability of the ligand within the binding groove across a comprehensive 230ns time scale, showing its therapeutic efficiency. The best pharmacophore was developed by using HipHop and was employed to screen a large database for the new hits over the best pharmacophore 18. SAR studies show that (compound 5a,b,c) 2- phenyl benzimidazole with an amide linkage to various substituted benzoyl chlorides shows a significant fit value of 4.029 in virtual screening methods. In vivo studies, the unsubstituted compound was selected as the prototype molecule. The compound does not show any toxicity up to 2000mg/ kg, and the remaining derivatives were tested against diclofenac as a positive control. SAR stated that halogen, electron-withdrawing functional groups at the 4th position of the terminal phenyl ring attached to the amide functionality were vital in augmenting the anti-inflammatory potential.
Compound 5(a)
Compound 5(b)
Compound 5(c)
Saikat Kumar Poddar et al. (2025) 2- substituted benzimidazole derivatives and evaluated the analgesic, anti-inflammatory, and antidiarrheal action by in silico and in vivo studies. The compounds were docked against diclofenac sodium (PDB ID- 1EQG and 5IKT) (−8.2 and −8.4 kcal/mol) as a standard drug; compound 6 shows the highest binding affinity in COX-1 and COX-2 (−8.7 and −8.9 kcal/mol, respectively). Cyclooxygenase (COX-1 and COX-2) plays an important role in prostaglandin and thromboxane synthesis 19,20. Prostaglandins are involved in inflammatory reactions, gastrointestinal cryoprotection, haemostasis and thrombosis, and renal haemodynamic. Therefore, these two enzyme isoforms were chosen to assess whether the in vivo analgesic and anti-inflammatory activities could result from inhibition of these enzymes by the synthesized compounds 21. Compounds 6(a) and 6(b) show the most prominent activity in biological evaluation and docking studies. These compounds exhibited the most potent anti-inflammatory activity in 1st hour to 4th hour (76.36, 76.60,71.43, 73.29, and 80.61, 88.30, 80.52, 66.07% inhibition of paw edema, respectively) compared to standard diclofenac sodium (56.97, 78.72, 90.91, and 99.64% inhibition of paw edema). Several benzimidazole derivatives have been reported to exert their anti-inflammatory activity by acting on different proteins and receptors, like COX enzyme, 5-lipoxygenase activating protein (FLAP), transient receptor potential vanilloid-1 (TRPV-1) ion channels, bradykinin receptors, cannabinoid receptors, and specific cytokines 22.
Compound 6(a)
Compound 6(b)
Azize Gizem Ergul et al (2024) investigated novel benzimidazole derivatives as potent inhibitors of microsomal prostaglandin E2 synthase 1, a promising target for treating inflammatory diseases and pain. Compound 7 was found to be the most potent analogue, exhibiting IC50 values of 0.27−7.0 nM in a cell-free assay for prostaglandin (PG)E2 production. This compound shows remarkable selectivity for PGES-1(IC50 = 2.9 nM) over COX-1, COX-2, 5-LOX, and FLAP, along with excellent bioavailability. COX-1 and COX-2 pathways show significant efficacy in reducing inflammation, but their prolonged usage can lead to severe GI and cardiovascular adverse effects. Selective inhibition of microsomal prostaglandin E2 synthase 1 (mPGES-1)-mediated PGE2 formation has emerged as an alternative target to these NSAIDs by inhibiting proinflammatory PGE2. The inhibition of mPGES-1 has demonstrated therapeutic efficacy over inflammation and anticancer types 23. The benzimidazole derivatives are developed to target opening new antiinflammatory agents that specifically target 5-lipoxygenaseactivating protein (FLAP), soluble epoxide hydrolase (sEH), and mPGES-1 within the AA pathway 24. In in vivo studies, the compound 7 reduces pain, inflammation, and fever significantly. At 30 mg/kg provides analgesic activity when compared to standard indomethacin. Toxicity studies indicate only mild alteration in liver enzyme markers compared to the standard drug, indicating increased selectivity and therapeutic efficiency for compound 7 25.
Compound 7
5. Integrated Inflammation–Cancer Signalling Pathway
Inflammation in the body can be triggered by internal (stress, aging) and external stimuli (chemicals, biological infections) 26. These inflammatory responses may trigger redness, swelling, pain, and dysfunction. Inflammatory cytokines such as TNF-α, IL-6, and IL-23 activate key oncogenic pathways, including NF-κB and STAT3, reinforcing a pro-tumorigenic inflammatory loop 28. The figure shows how inflammation triggers tumour initiation.
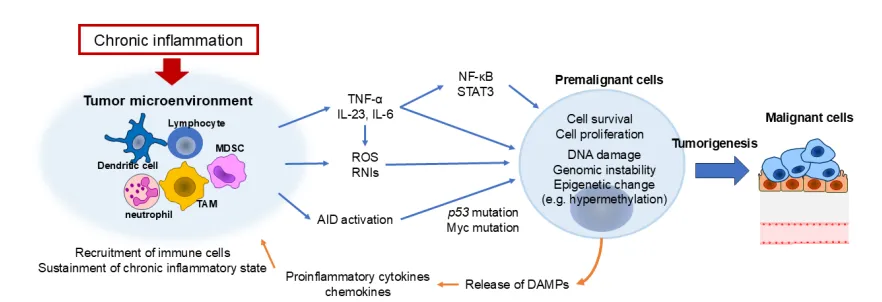
FIGURE 2 | Inflammation triggers tumour initiation
Tara Williamson et al. (2016) developed a chemopreventive strategy combining an NSAID (Sulindac) with an anthelmintic drug containing benzimidazole (mebendazole). This oral drug combination was effective in the ApcMin/+ mouse model of Familial Adenomatous Polyposis (FAP). Treatment with 35 mg/kg daily mebendazole reduced the number of intestinal adenomas by 56% (P = 0.0002), 160 ppm sulindac by 74% (P < 0.0001), and the combination by 90% (P < 0.0001) 27. Here, the mebendazole reduces COX-2 expression, blood vessel formation, kinase activity, and reduces the secretion of pro-inflammatory cytokines. Katarzyna Błaszczak-Swi ˛atkiewicz et al. (2019) studied the antiproliferative activity of 2 substituted benzimidazole derivatives by inhibiting NF-κB expression. The activity of the tested compounds on T47D and MCF7 cells was examined by WST, western blot, NF-κB transactivation assay, and apoptotic cell population analysis 29. Compound 8 shows the most potent activity with an IC50 of 0.31 ± 0.06 nM, although weaker than tirapazamine, and was significantly higher than the other tested compounds (2.4–3.0 fold). This compound appears to be the strongest inhibitor of NF-κB expression in hypoxic conditions.
Compound 8
Reshma Sathyanarayana et al. (2021) synthesised benzimidazole derivatives with varied carbon chain length through one-pot nitro reductive cyclization and evaluated their anticancer and anti-inflammatory activity. Among the derivative compounds, 9 (a) shows anticancer activity by maximum cell death in HeLa and A549 cell lines, and compound 9(b) emerged as a potent anti-inflammatory agent through in vivo studies (carrageenan-induced paw edema method). 9(a) having 4-Cl substitution and methyl chain length with 64.31% inhibition. The inhibitory potency of these derivatives increased at the end of the 3rd hour, i.e., up to 77.50%, which was comparable with the reference drug indomethacin (74.02%) 30.
Compound 9(a)
Compound 9(b)
LIMITATIONS AND FUTURE PERSPECTIVE
Even though benzimidazole has enormous therapeutic potential, various hurdles and constraints must be overcome before its clinical applications can be completely realized. Limitations include the unwanted binding with the off target, resulting in unpleasant reactions and physiological toxicity. Un pleasant reactions or unwanted physiological responses. The potential for cytotoxicity, genotoxicity, and organ-specific toxicity must be extensively examined during preclinical research to determine the drug’s safety profiles and create optimal dose regimens 31.
Future perspectives include personalised medicine using benzimidazole by genome and biomarker identification. In future rational design strategies, integrating computational modelling, molecular docking, and advances in SAR activity studies to optimise potency and selectivity. Exploring Multitarget-directed ligands of benzimidazole, thereby targeting multifactorial diseases. Exploration of green chemistry and sustainable methodologies will expand the novel benzimidazole ligands. In general, the future of research on benzimidazole is focused on harnessing its chemical adaptability to meet clinical demands that have not yet been fulfilled, as well as providing therapeutics that are safer, more effective, and more user-friendly for patients.
REFERENCES


Sangeetha Nair, Seema Nair, Benzimidazole Scaffold as A Dual Modulator of Cancer and Inflammation -A Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 4166-4175, https://doi.org/10.5281/zenodo.20730388
 10.5281/zenodo.20730388
10.5281/zenodo.20730388