
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Sciences, RIMSR, Puthuppally, Kottayam, Kerala, India 686009
Because of the hostile gastrointestinal environment, which is marked by severe enzymatic degradation and poor mucosal permeability of hydrophilic macromolecules, oral administration of human insulin poses a significant biopharmaceutical challenge. However, by emulating the physiological portal insulin gradient and removing the drawbacks of subcutaneous injections, effective oral administration has the potential to completely transform the management of diabetes. In order to overcome these intricate biological obstacles, this paper thoroughly investigates the synergistic application of Hydrophobic Ion Pairing (HIP) and of Solid Supersaturable Self-Microemulsifying Drug Delivery Systems (S-SuSMEDDS) . By efficiently converting hydrophilic insulin into a lipophilic compound, the non-covalent HIP technique protects it from presystemic breakdown and permits maximum drug loading within the lipid matrix. Customized polymeric precipitation inhibitors are incorporated into Su-SMEDDS to address the thermodynamic instability of traditional microemulsions upon water dilution. These polymers maximize the intestinal absorption window and prevent premature crystallization by kinetically stabilizing insulin in a supersaturated state through the "spring and parachute" action. Additionally, using high-surface-area mesoporous adsorbents to convert liquid systems into solid dosage forms (S-SuSMEDDS) addresses important industrial issues like lipid oxidation and capsule shell incompatibility, guaranteeing a stable, free-flowing powder appropriate for conventional manufacturing. This review demonstrates the translational potential of S-SuSMEDDS by describing the molecular principles, specific characterisation methods, and in vivo performance measures. In the end, combining HIP complexation with solid supersaturable lipid substrates provides a significant, multifunctional paradigm shift toward the efficient, non-invasive oral delivery of therapeutic peptides.
Diabetes Mellitus
Diabetes mellitus (DM) is a metabolic disease involving inappropriately elevated blood glucose levels1. DM has several categories, including type 1, type 2, maturity-onset diabetes of the young (MODY), gestational diabetes, neonatal diabetes, and secondary causes due to endocrinopathies, steroid use, etc. The main subtypes of DM are Type 1 diabetes mellitus (T1DM) and Type 2 diabetes mellitus (T2DM), which classically result from defective insulin secretion (T1DM) and/or action (T2DM)1. T1DM presents in children or adolescents, while T2DM is thought to affect middle-aged and older adults who have prolonged hyperglycaemia due to poor lifestyle and dietary choices1. The pathogenesis for T1DM and T2DM is drastically different, and therefore each type has various etiologies, presentations, and treatments2.
The two primary categories of endocrine cells in the pancreatic islets of Langerhans are beta cells, which produce insulin, and alpha cells, which secrete glucagon. Depending on the glucose environment, beta and alpha cells constantly alter the amount of hormones they secrete3. The glucose levels become improperly imbalanced when insulin and glucagon are out of balance4. The loss of beta cells in the pancreas, usually as a result of an autoimmune disease, is the hallmark of type 1 diabetes. Insulin is either completely absent or very low as a result of the complete death of beta cells. This causes an imbalance between insulin levels and insulin sensitivity, resulting in a functional insulin deficit in type 2 diabetes, which has a more subtle beginning4.
Insulin Therapy in Diabetes Mellitus
The management of diabetes mellitus has been inherently transformed by the evolution of insulin therapy, which remains a cornerstone of glycaemic control for millions in worldwide. In Type 1 Diabetes Mellitus (T1DM), the absolute insulin deficiency prevails due to autoimmune destruction of the pancreatic beta-cell. The exogenous insulin is a life-sustaining necessity, typically administered through intensive basal-bolus regimens or continuous subcutaneous infusions5. In contrast to Type 2 Diabetes Mellitus (T2DM), insulin is strategically introduced due to progressive beta-cell exhaustion, which renders oral antidiabetic agents and lifestyle modifications insufficient to meet glycaemic targets6. The modern clinical practice has shifted significantly towards insulin analogues, which are variants of human insulin7. They offer superior pharmacokinetic and pharmacodynamic profiles. The rapid-acting analogues provide more precise postprandial coverage, while long-acting, peakless basal analogues significantly reduce the risk of nocturnal hypoglycaemia compared to traditional NPH insulin. Since they closely mimic endogenous physiological secretion, these analogues enhance both metabolic stability and patient quality of life, addressing the diverse therapeutic needs across the diabetic spectrum1.
Human Insulin
Human insulin is a 51-amino-acid, 5.7-kDa protein naturally produced by pancreatic islet beta cells, making it one of the most thoroughly studied proteins in medicine5. Structurally, the insulin molecule is comprised of an A chain and a B chain that are chemically linked by two di-sulphide bridges. At the cellular level, this hormone is synthesized and stored within the vesicles of the Golgi-apparatus as hexamers—a stabilized structural form resulting from high local insulin concentrations and the presence of zinc4.
Mechanism of Action Of Insulin
Human insulin is instrumental in maintaining systemic glucose, lipid, and energy homeostasis7. Following its dissociation into biologically active monomers or dimers, its physiological effects are primarily exerted within skeletal muscle, adipose tissue, and the liver via binding to the insulin receptor (IR). The IR functions as a heterotetrameric tyrosine kinase, structured with two extracellular α-subunits and two intracellular, catalytic β-subunits. Ligand binding precipitates a conformational shift in the β-subunits, triggering their intrinsic tyrosine kinase activity and subsequent autophosphorylation. This molecular event facilitates the recruitment of docking proteins, initiating a signalling cascade through phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) and protein kinase B (Akt). Ultimately, Akt serves as a central regulator of glucose transporter type 4 (GLUT4), protein kinase C (PKC), and mitogen-activated protein kinase (MAPK) pathways4.
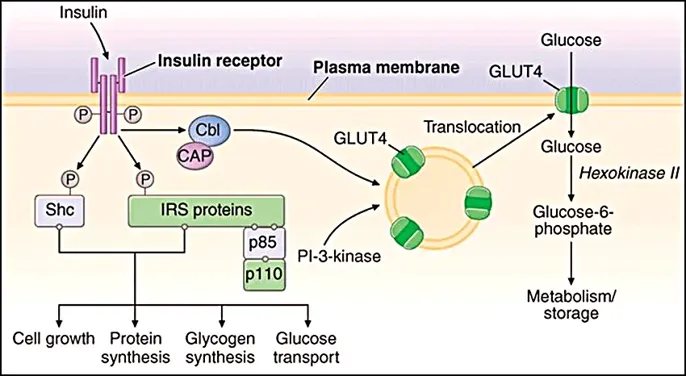
Fig.No.1. Mechanism of action of Insulin8
Disadvantages Of Injectable Delivery
The administration of insulin glargine via the injectable route presents several clinical and patient-related challenges1. Primarily, the necessity of frequent injections leads to significant injection-site pain, localized tissue injury, and the development of lipodystrophy, which can impair the absorption consistency5. From a physiological standpoint, injectable delivery bypasses the natural portal-arterial insulin gradient, which results in peripheral hyperinsulinemia2. This abnormal distribution increases the risk of hypoglycaemic events and can promote weight gain through systemic anabolic effects5. Moreover, this invasive delivery method necessitates a high degree of patient compliance and technical skill, often leading to treatment non-adherence, needle phobia, and an increased burden on healthcare resources for the management of administration-related complications2.
The Barriers of Oral Insulin Delivery
Various non-invasive methods for the administration of insulin, including oral, transdermal, pulmonary, ocular, and vaginal pathways, have undergone rigorous investigation. Regardless of these efforts, achieving therapeutic bioavailability remains a significant hurdle across the different delivery routes. Among them, the oral route is prioritized due to superior patient compliance and ease of administration. From a physiological perspective, the oral delivery of insulin which closely reflects endogenous secretion by utilizing the portal circulation5. Thus, mitigating the risk of hypoglycaemia, which is associated with subcutaneous insulin injections. In addition to the evidence that this route may actively contribute to the preservation and maintenance of pancreatic β-cell functions9.
Though the gastrointestinal (GI) tract is dedicated to the digestion of food, nutrient absorption, and defence against outside pathogens, it has a complex environment in terms of its physiological structures and chemical components. The biochemical barriers encountered when delivering therapeutic peptides and proteins orally mainly consist of acidic pH levels, a variety of enzymes, surfactants, and both exogenous and endogenous thiols. The luminal pH of the entire gastrointestinal tract varies considerably from the stomach to the end of the small intestine. Generally, the pH of gastric fluid falls between 1 and 2, and there is a significant shift from high acidic pH in the stomach to an almost neutral pH level in the duodenum (6-6.5), and increasing to a pH range of 7 to 8 in the terminal ileum10.
Self-Microemulsifying Drug Delivery Systems (SMEDDS)
Self-microemulsifying drug delivery systems (SMEDDS) are defined as physically stable, isotropic mixtures consisting of a lipid phase (oils), surfactants, and co-surfactants, in which the active pharmaceutical ingredient (API) is fully solubilized. Upon oral administration, these systems will use the natural peristaltic motility of the stomach and small intestine as a gentle agitation. This interaction with the aqueous gastrointestinal environment triggers the spontaneous formation of transparent, stable, fine oil-in-water (o/w) emulsion (< 50nm)11. By keeping the drug in a pre-dissolved state within a microscopic oil droplet, SMEDDS bypass the traditional dissolution challenges associated with hydrophobic drugs, thereby enhancing their oral bioavailability12.
The successful development of Self-Micro Emulsifying Drug Delivery Systems (SMEDDS) depends on the strategic screening and selection of the lipid phase, surfactants, and co-surfactants. The relative ratios of these three components dictate the resulting globule size upon aqueous dilution; specifically, SEDDS will typically form conventional emulsions, whereas SMEDDS generate transparent and more thermodynamically stable microemulsions. To determine the optimal concentration ranges required for spontaneous emulsification, researchers apply pseudo-ternary phase diagrams. These diagrams are crucial for defining the "self-emulsification region" and assessing how drug loading influences the stability and formation of the microemulsion. A defining characteristic of SMEDDS is a mean globule size of less than 50 nm. This ultra-fine dispersion provides a significantly expanded interfacial surface area, which serves as a critical driver for rapid drug release and enhanced intestinal absorption13. In liquid SMEDDS, the therapeutic agent is maintained in a solubilized state within the lipid phase, commonly encapsulated in either soft or hard gelatin capsules for oral administration14.
Thermodynamic Principle of SMEDDS
A significant limitation of conventional emulsions is their inherent thermodynamic instability; as metastable dispersed systems, they are highly sensitive to environmental changes and typically require high-shear agitation for formation14,15. In contrast, SMEDDS are physically stable and form spontaneously. The spontaneous nature of the emulsification process is governed by the change in Gibbs free energy (ΔG) of the system:
ΔG=ΔH-TΔS.
ΔH represents the change in enthalpy of the system, corresponding to the energy required to expand the interfacial surface area. The change in entropy (ΔS) represents the extent of size reduction of the oil phase and the subsequent exponential increase in the total number of dispersed droplets. Emulsification proceeds when the positive entropy change (ΔS), which drives the dispersion of the phase, exceeds the enthalpic energy (ΔH) required to expand the interfacial surface area11,16. According to the Reiss equation, the free energy changes necessary to achieve a stable microemulsified state upon mild agitation in the gastrointestinal tract11.
(ΔG = Σ Ni π r²iσ)
ΔG = Change in Gibbs energy
Ni = Total number of oil droplets
r = Droplet radius
σ = Interfacial tension
In a conventional emulsion system, the high interfacial tension (σ) causes a massive positive enthalpy, requiring mechanical energy to induce emulsification. However, the inclusion of surfactants and co-surfactants reduces the interfacial tension (σ) to near zero value. So the (σ)is too low, the energy required to expand the interface is drastically minimized. This reduction allows the mass positive entropy (ΔS) of droplet dispersion to dominate the equation, which results in a negative overall Gibbs free energy11. This thermodynamic shift is what allows the formulation to achieve a stable microemulsified state using only mild agitation provided by GI motility10.
Supersaturatable SMEDDS (Su-SMEDDS)
The performance of a SMEDDS formulation depends on its ability to prevent drug precipitation following aqueous dilution in the GI tract. While the inclusion of elevated surfactant levels can stabilize the drug in a dissolved state, this approach is often limited by the potential for surfactant-induced GI toxicity14,15. To rectify this, researchers developed Supersaturatable Self-Microemulsifying Drug Delivery Systems (Su-SMEDDS). This methodology involves incorporating a polymeric precipitation inhibitor to maintain a metastable supersaturated state upon dilution17. The primary mechanism of PI polymer chains will adsorb onto the surfaces of nascent nuclei or growing crystals. This will sterically hinder both nucleation and crystal growth. The dual-action strategy is widely described as the “spring and parachute” effect12,17–21.
Precipitation Inhibitors
A precipitation inhibitor is defined as a pharmaceutical excipient which maybe a polymer or a surfactant incorporated into a lipid formulation to delay the thermodynamically or kinetically induced crystallization and precipitation of poorly water-soluble drugs which forms a dispersion upon dilution with Gi fluids18.
Mechanism Of Action of PIs
The moa of PIs involves a multi-pathway process that stabilizes supersaturate solutions by inhibiting both nucleation and crystal growth phases. Hydrophilic polymers, primarily delay the onset of precipitation by forming strong intermolecular contacts with drug molecules through hydrogen bonding. simultaneously, the hydrophobic characteristics of these polymers will allow their direct adsorption to the drug's crystal surfaces, forming a physical barrier that will stop the crystalline growth. The cellulosic polymers swell to create a wide reticulate network of entangled structural chains when comes in contact with the aqueous phase. This entangled polymer web imposes considerable steric hindrance against crystallization and enhances localized viscosity, especially when high polymer weight grades are used. Finally, this viscous barrier ensures that the formulation stays in a kinetically stable, supersaturated condition for optimal absorption by rigidly limiting the diffusion rate of drug molecules from the bulk solution to the crystal nucleus9.
Common examples include18,20;
Cellulose derivatives- Hydroxypropyl Methyl Cellulose, Hydroxypropyl Cellulose.
Vinyl polymers- Polyvinylpyrrolidone, polyvinyl alcohol,
Amphiphilic co-polymers- Soluplus®
Cyclodextrins- Hydroxypropyl-β-cyclodextrin
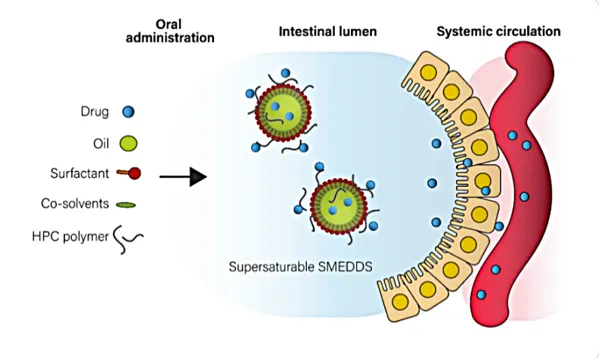
Fig.No.2. Mechanism of action of PI’s22
Advantages of Su-SMEDDS
1. Utilizes polymer-based precipitation inhibitors to maintain drugs in a kinetically stable, supersaturated state.
2. Achieves significantly higher drug loading than conventional formulations because they stabilize the drug at concentrations above its equilibrium solubility.
3. Maintains a stable state with lower surfactant levels (often reducing the conventional requirement of >30% w/w)17.
Solid-SMEDDS and Adsorbents
Liquid formulations often have incompatibility problems with capsule shells, such as leakage or softening. To resolve these challenges, Solid-SMEDDS (S-SMEDDS) have been introduced14. In S-SMEDDS, a solid is an adsorbent which are highly porous, pharmacologically inert materials characterized by high surface area and large porous volume, the main function is to transform liquid or semi-solid formulations into a stable, free-flowing powder via the adsorption technique. This allows converting into unit dosage forms like tablets or capsules12,21,23–25. A Niche carrier will maintain the drug in an amorphous state, preventing the recrystallization of poorly water-soluble drugs and thereby enhancing their dissolution and stability23.
Table.1.Types of Solid Adsorbents26,27
|
Solid Carrier / Trade Name |
Chemical Composition |
Specific Surface Area (m2/g) |
Average Pore Volume / Size |
Key Formulation Characteristic |
|
Aerosil® 200 |
Colloidal Silicon Dioxide (Fumed) |
200 ± 25 |
High external area (non-porous) |
Rapid oil adsorption; acts as an excellent glidant for powder flowability. |
|
Syloid® 244 FP |
Amorphous Silica Gel |
~300 |
Mesoporous (~1.6 m2/g) |
Maximum liquid loading capacity due to a vast internal porous network. |
|
Neusilin® US2 |
Magnesium Aluminometasilicate |
~300 |
Mesoporous (~1.0 m2/g) |
Exceptional direct compressibility; protects sensitive lipids from oxidation. |
|
Aeroperl® 300 |
Granulated Colloidal Silicon Dioxide |
260 – 320 |
Mesoporous (~1.6 m2/g) |
Dust-free handling with high intra-particle porosity for liquid entrapment. |
|
Fujicalin® |
Dibasic Calcium Phosphate Anhydrous |
30 – 40 |
Macroporous (Mean pore: ~7 nm) |
Moderate oil adsorption but provides outstanding mechanical tablet strength. |
Mechanism of adsorption in Solid-Su-SMEDDS
The mechanism of adsorption mainly relies on the physical interaction of liquid Su-SMEDDS and the micro-architectural properties of solid carriers. The highly porous adsorbents, such as Aerosil® 200, Neusilin®, are liquid formulation is drawn into the carrier’s porous network by robust capillary action and surface adsorption12,24. As long as in this physical entrapment, the solubilized active ingredient is confined to a stable, amorphous state within the solid matrix. The highly porous materials maximize the lipid-loading capacity without forming irreversible chemical bonds between carriers and formulation. Thus ensuring a stabilized formulation which will rapidly desorb and be effectively released upon introduction to the aqueous GI environment25.
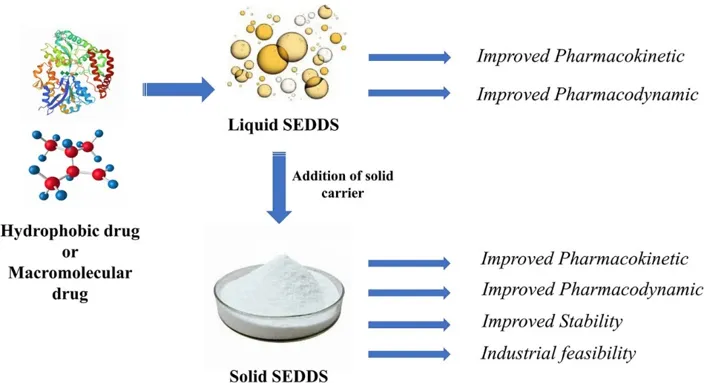
Fig.No.3. Mechanism of adsorption in Solid-Su-SMEDDS28
Lipidization and Covalent Strategies
In order to administer therapeutic peptides and proteins, lipidization techniques are essential to enhance the lipophilic character of hydrophilic macromolecules. Generally, for the lipidization, non-covalent and covalent methods are used29.
In the noncovalent methods, the hydrophobic Ion Pairing method strategy effectively improves the lipophilicity of peptides, protecting against presystemic enzymatic degradation and enhancing intestinal membrane permeability29.
The hydrogen bond is a dipole-dipole interaction due to the covalent bonding between a hydrogen atom and a highly negative atom like N2or O2 in peptides. H2 bond pairing improves the lipophilicity of peptides by forming intermolecular and intramolecular bonds between nonionic surfactants. This H2 bonding will give necessary lipophilicity, cage protection from enzymatic degradation, and enhance the membrane permeation29.
The reverse micelle technique encapsulates hydrophilic peptides within the protective aqueous core of a water-in-oil microemulsion. This non-covalent shielding protects the drug from enzymatic degradation and the surrounding oil phase, enabling high-payload integration into lipid systems like S-SMEDDS while fully preserving the peptide's native structure and biological activity6,30–32.
In the covalent strategy, the prodrug technique converts active therapeutic drugs into inactive precursors to optimize physicochemical properties. Lipophilic moieties are conjugated to the functional group peptide (e.g., amino, carboxyl, or hydroxyl groups). Reversible covalent lipidization allows for improved absorption without risking permanent loss of biological activity29.
Hydrophobic Ion Pairing (HIP) Method
Hydrophobic Ion Pairing (HIP) has developed into a key non-covalent method for turning hydrophilic, charged drugs into lipophilic complexes29,33,34. It is based on ionic interactions between an oppositely charged counterion with hydrophobic areas and a polar drug molecule29. The successful construction of a robust hydrophobic ion pair depends on selecting appropriate hydrophobic counterions:
- Anionic Counterions: Carboxylates (e.g., sodium oleate, sodium deoxycholate), phosphates, sulfonates, and sulfates (e.g., sodium docusate, sodium dodecyl sulfate). These pair effectively with cationic ammonium substructures found on peptides34.
- Cationic Counterions: Quaternary amines and alkylamines pair with anionic groups34.
- Divalent Metal Ions: Ca2+, Zn2+, and Cu2+ serve as effective alternatives. For example, Zn2+ is extensively utilized to engineer water-insoluble zinc–peptide complexes like insulin and exenatide5. While HIP masks the peptide’s hydrophilic residues, increasing its Log P, it decreases aqueous solubility. Thus, the stoichiometric ratio of drug-to-counterion must be precisely optimized34.
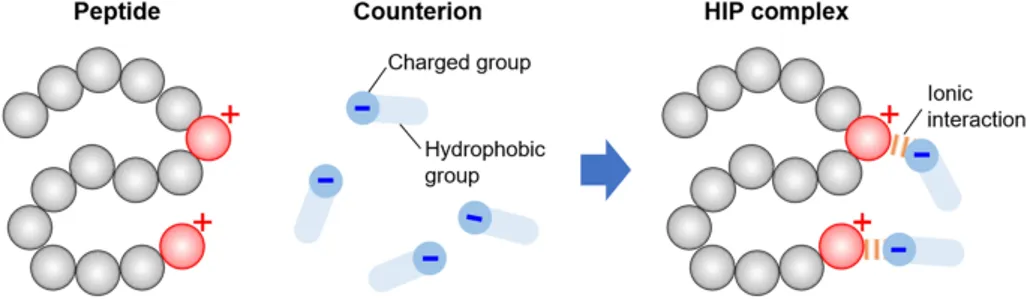
Fig.No.4. HIP Method35
Ingredients For SU-SMEDDS
Lipid phase: Lipids are fatty acids and their derivatives, which are amphiphilic in nature due to their dual molecular structure. A lipophilic portion consisting of fatty acid(s) and the hydrophilic portion, in which are the esterified fatty acids11.
The oil phase will form a spontaneous microemulsion, enhance solubility, and provide a fraction of the lipophilic drug transported through the intestinal lymphatic system. Medium-chain triglycerides (MCTs) are considered due to their superior solvent properties and enhanced resistance to oxidative degradation compared to LCTs (Large-Chain Triglycerides)11.
(MCTs, e.g., palm seed, coconut oil, Capmul MCM.
LCTs, e.g., corn, olive, peanut, rapeseed, sesame or soybean oils
Surfactants
With an aim to achieve high emulsifying properties, emulsifiers with high HLB and high hydrophilicity should be used. By reducing the interfacial tension between oil and water, this leads to the spontaneous formation of oil-in-water microemulsion and the rapid dispersion of the formulation in aqueous media. Non-ionic surfactants having high Hydrophilic-Lipophilic Balance (HLB) values are opted for their safety profiles and stabilizing abilities11,36.
Co-Surfactants
Co-surfactants are used in SMEDDS to increase drug loading, improve self-emulsification time, and modulate microemulsion droplet size. These are achieved by modulating the mechanical properties of the interfacial film. HLB values between 10 and 14, they could increase the fluidity and elasticity of the interface, thus effectively lowering the interfacial tension required for emulsification12.
Precipitation Inhibitors (PI):
PI acts as the essential "parachute" for the formulation, having hydrophilic polymers, which temporarily block nucleation and crystal growth through steric hindrance and intermolecular hydrogen bonding. These two mechanisms will effectively prolong the window of supersaturation, preventing the drug from crashing out of solution and ensuring the dissolved drug is long enough to be fully absorbed. Common polymeric PIs incorporated into these systems include Hypromellose (HPMC), Soluplus®, and Polyvinylpyrrolidone (PVP)9,18,20.
Characterization And Evaluation
Evaluation of Ins-Counterion Complex
The test is used to determine how successfully the insulin is complexed with the counterion to form a neutral, lipophilic, and water-soluble complex. For the test, the supernatant is analysed via HPLC after mixing and centrifugation to quantify the unbound peptide7,20,31.
This test signifies the change in lipophilicity of the drug. The n-octanol/water partition coefficient is measured. A shift to a positive Log P value is the primary indicator of successful Hydrophobic Ion Pairing (HIP) complexation7.
This test signifies the maximum solubility of the complex in water. The solubility of the complex in water is drastically reduced if the complex is successfully formed, when compared to the native insulin. Also, the solubility study is carried out in the oil phase, which shows an increased solubility in the oil phase7.
Fourier Transform Infrared Spectroscopy is used to validate the structural integrity of the complex formed. This will confirm the complexation through characteristic shifts in vibrational frequencies while ensuring the essential Amide bands remain unchanged2.
Differential Scanning Calorimetry is a thermal analysis technique that is utilized to assess the physical state of the formulation components. This will specifically confirm the successful transition of the active ingredient into an amorphous state12.
The test is to visually confirm the morphological shift. That is native insulin often appears as geometrical crystals, whereas the HIP complex is typically seen as irregular, aggregated, sponge-like, or amorphous masses11.
This spectroscopic method evaluates the conformational stability of the peptide structure of inulin. Monitoring specific wavelengths at 208 nm and 222 nm, it ensures that insulin's critical alpha-helical structure is unchanged even after formulation11.
(Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis) Used to confirm there is no covalent aggregation, fragmentation, or irreversible chemical degradation of the peptide chain during the precipitation and drying phases34.
This study is conducted to verify that the drug can be effectively released from the complex once entered in the blood circulation. It confirms the necessary reversibility of the HIP complex when reached in physiological media34.
Evaluations Of Liquid SU-SMEDSS
This test is evaluated by diluting the formulation in an aqueous medium and observing its appearance. A clear and transparent dispersion indicates the successful formation of a microemulsion, whereas an opaque or turbid appearance signifies a macroemulsion12.
These nanometric parameters are quantified using Dynamic Light Scattering (DLS) to ensure optimal microemulsion characteristics. For effective systemic delivery, the target metrics are a Z-average droplet size of less than 100 nm, a Polydispersity Index (PDI) below 0.3, and an absolute Zeta Potential greater than ±30 mV for colloidal stability11.
This test involves testing the exact time in seconds taken by the formulation to form a homogeneous microemulsion by gentle stirring. And it is graded from I to V11.
A transmittance test evaluates the clarity of the diluted formulation using a UV-Vis spectrophotometer. A high transmittance value (greater than 95%) is typically required to confirm the formation of a fine, clear microemulsion12.
This test confirms that the liquid preconcentrate is not too viscous, so that they easily disperse and forms a microemulsion upon contact with GI fluid26.
TEM with negative staining is used to visualize the morphological shape and structural integrity of droplets produced12.
This evaluation will study the physical robustness of the formulation when subjected to environmental stressors. It is rigorously assessed through a series of heating-cooling cycles, centrifugation, and freeze-thaw tests to confirm the absence of phase separation or drug precipitation12.
Performance And In-vivo Studies
The PI test evaluates the formulation's ability to maintain drug supersaturation upon aqueous dilution, monitoring the essential "spring and parachute" effect. It is conducted in simulated gastrointestinal fluids to ensure the incorporated precipitation inhibitors effectively prevent drug crystallization9.
The release kinetics of the API from the formulation are systematically assessed across various pH media that mimic the environment of the gastrointestinal tract. This release profile is determined by using either dialysis bag techniques or the standard USP dissolution apparatuses9.
This assay determines the capability of formulation to shield fragile peptide drugs from premature presystemic degradation. The test is evaluated by incubating the delivery system with key gastric and intestinal digestive enzymes, such as pepsin, trypsin, and chymotrypsin3,32.
This study signifies the formulation's capacity to enhance intestinal absorption across the mucosal barrier. Transport efficiency is quantified by the everted rat gut sac model to accurately determine the apparent permeability coefficient3.
This in vivo evaluation test confirms the actual therapeutic efficacy of the formulated delivery system.
It is conducted using diabetic rat models, which are induced by streptozotocin, to validate the drug's sustained hypoglycaemic effects and overall systemic bioactivity1.
Evaluation Of Capsule Formulation
This test includes proper checking for uniform colour, proper locking of the cap and body, and the absence of surface defects. And also look for shell softening, deformation, or spotting, which indicates that the oil phase is interacting with or leaking through the capsule shell14.
This test involves weighing the filled capsules, as well as weighing the contents and capsule shells after emptying the shells. The weight variation should be within the specified limit mentioned in the IP/USP.
In this test, the amount of drug in a capsule is identified. Random capsule samples are used to identify the amount of drug in it by the HPLC method. The content variability should be in a strict percentage range of the label claim26.
Capsules are placed in a standard USP disintegration apparatus, and the disintegration time is determined using simulated gastric fluid. This shows the exact time for formulation release and also states the effect of the capsule shell on the release of the formulation17.
The test is carried out using USP apparatus I (basket) or apparatus II (paddle). It shows the drug release study over time and also the supersaturation effect of formulation17.
CONCLUSION
One of the most difficult problems in modern biopharmaceutics is the oral administration of biological macromolecules, especially human insulin, because of the gastrointestinal tract's hostile enzymatic environment and the poor mucosal permeability of hydrophilic peptides. The integration of Solid Supersaturable Self-Microemulsifying Drug Delivery Systems (S-SuSMEDDS) with the Hydrophobic Ion Pairing (HIP) technology provides a highly synergistic, multifunctional strategy to overcome these obstacles, as described in this paper.
The naturally hydrophilic insulin molecule is successfully converted into a highly lipophilic complex by using the HIP technique. This non-covalent alteration offers a crucial steric barrier against presystemic enzymatic degradation in addition to enabling substantial drug loading within the lipid phase. The ultra-fine lipid droplets further improve membrane penetration and lymphatic transport after spontaneous microemulsification in the gastrointestinal fluids. Additionally, the thermodynamic weaknesses of traditional SMEDDS are addressed by the use of certain polymeric precipitation inhibitors (PIs). These polymers prevent nucleation and crystal formation by taking advantage of the "spring and parachute" effect, which keeps the insulin in a kinetically stable, supersaturated state long enough to optimize the intestinal absorption window. Furthermore, the practical and industrial constraints of lipid-based formulations, such as capsule shell incompatibility, lipid oxidation, and drug leakage, are resolved by the conversion of liquid Su-SMEDDS into solid dosage forms via high-surface-area mesoporous adsorbents. In addition to providing the flowability and compressibility needed for the large-scale production of tablets or capsules, the physical entrapment of the microemulsion within these porous designs keeps the complex in an amorphous, extremely stable condition.
FUTURE PERSPECTIVE
Although HIP-driven S-SuSMEDDS's mechanistic justification and in vitro performance are very convincing, thorough in vivo pharmacokinetic (PK) and pharmacodynamic (PD) profiling in higher-order mammalian models will be necessary for the platforms' effective clinical translation. The long-term toxicological effects of repeated exposure to high concentrations of surfactants, precipitation inhibitors, and complexing counterions on the gastrointestinal mucosa must be the main focus of future research. Furthermore, commercial scalability will depend on improvements in continuous production techniques like spray-drying or hot-melt extrusion of these solid-lipid systems. In the end, the S-SuSMEDDS platform represents a significant paradigm change in the delivery of peptides. It has the potential to significantly enhance therapeutic compliance and quality of life for millions of people managing diabetes worldwide, successfully replace invasive subcutaneous regimens, and closely resemble the physiological portal insulin gradient.
REFERENCES





Anandhu Dineshan, Anuroop UP, Arun Raj R, Akhila P, Nebin Koshy, Bridging the Oral Peptide Delivery Gap: Hydrophobic Ion Pairing and Solid Supersaturable SMEDDS for Human Insulin- A Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 7460-7474. https://doi.org/10.5281/zenodo.21054023
 10.5281/zenodo.21054023
10.5281/zenodo.21054023