
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, S. N. College of Pharmacy, Jaunpur, Uttar Pradesh
Non-steroidal anti-inflammatory drugs (NSAIDs) like Aceclofenac represent a cornerstone in the pharmacological management of chronic inflammatory and musculoskeletal conditions, including osteoarthritis and rheumatoid arthritis. While Aceclofenac effectively alleviates pain and inflammation through the inhibition of cyclooxygenase (COX) enzymes, its clinical utility is frequently compromised by severe, dose-limiting gastrointestinal (GI) toxicity. This localized mucosal damage and ulcerogenesis are primarily driven by the drug's free terminal carboxylic acid moiety, which induces direct cellular irritation and toxic intracellular ion trapping within the acidic environment of the stomach. To circumvent this therapeutic liability, the present research aimed to rationally design and synthesize a novel, gastro-sparing Aceclofenac prodrug derivative by transiently masking the offending acidic functional group via a reactive haloacetic ester intermediate. The structural modification was strategically executed utilizing a rational prodrug paradigm. In the preliminary phase, the essential haloacetic ester intermediate, ethyl 2-bromoacetate, was synthesized via a classical acid-catalyzed Fischer esterification. Subsequently, the final Aceclofenac ester derivative, ethyl 2-[[2-[2-[2-[(2,6-dichlorophenyl) amino] phenyl]acetyl]oxyacetyl]oxy]acetate, was successfully synthesized through a bimolecular nucleophilic substitution (SN2) reaction. This involved the basic deprotonation of Aceclofenac using anhydrous potassium carbonate in a polar aprotic solvent (N, N-Dimethylformamide), followed by coupling with the electrophilic ?-carbon of the intermediate. The synthesised derivative emerged out as a dark powder with a melting point range of 144–160°C and a yield of 59.2%. The physicochemical assessment validated that the masking of the carboxylic acid markedly enhanced the lipophilicity of the parent molecule, making it virtually insoluble in water while exhibiting high solubility in organic solvents like dichloromethane, chloroform, and N,N-Dimethylformamide. Complete spectroscopic characterisation definitively confirmed the chemical structure of the synthesised derivative. Fourier Transform Infrared (FT-IR) spectroscopy validated the successful esterification by the emergence of specific ester carbonyl (C=O) stretching bands at 1752 and 1735 cm-1, with the absence of the wide carboxylic acid O-H stretch. Mass spectrometry (ESI+) confirmed the structure even more by showing a main [M+H]+ molecular ion peak at m/z 441.1, a typical [M+Na]+ sodium adduct at m/z 463.1, and predicted fragmentation patterns. 1H-NMR spectroscopy also gave clear evidence of the terminal change, showing the particular breaking patterns of the new ethyl ester tail: a methylene quartet at ? 4.15 (2H) and a terminal methyl triplet at ? 1.20 (3H). UV-Visible spectroscopy (???? 275 nm) verified that the fundamental diphenylamine core pharmacophore necessary for COX binding remained entirely intact and structurally unaltered. Structure-Activity Relationship (SAR) insights derived from this study highlight that this terminal esterification effectively eliminates direct acidic contact with the gastric mucosa while pushing the volumetric limits to enhance the compound's partition coefficient (LogP). This transient, biolabile ester acts as a protective shield, allowing the highly lipophilic prodrug to safely bypass the stomach's acidic environment while potentially improving passive cellular permeability. Once systemically absorbed, it is hypothesized to undergo rapid hydrolysis by ubiquitous tissue and plasma esterases, safely releasing the active parent drug. In conclusion, this research establishes a comprehensive synthetic framework for the evolution of the Aceclofenac scaffold, delivering a rationally designed candidate that holds significant promise as a metabolically robust, non-ulcerogenic anti-inflammatory therapeutic.
1.1 Background on Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
Inflammation is a basic biological response to harmful stimuli, tissue damage, or infection. It is made up of a complicated series of biochemical and cellular activities. Non-steroidal anti-inflammatory medications (NSAIDs) are one of the most commonly prescribed types of drugs in the world. They are mostly used for their strong pain-relieving, fever-reducing, and anti-inflammatory effects. They are the mainstay of symptomatic treatment for a wide range of clinical illnesses, from acute postoperative pain to chronic, debilitating inflammatory disorders such rheumatoid arthritis, osteoarthritis, and ankylosing spondylitis [1].
The main way that conventional NSAIDs work is by stopping the cyclooxygenase (COX) enzyme system, which stops the arachidonic acid cascade. NSAIDs stop the production of downstream eicosanoids, especially pro-inflammatory prostaglandins (PGs), prostacyclins, and thromboxanes, by inhibiting COX [2]. There are two main types of the COX enzyme: COX-1 and COX-2. In most healthy tissues, COX-1 is an enzyme that is always present. It is a "housekeeping" enzyme that is very important for keeping proper physiological homeostasis. This includes controlling renal haemodynamics, platelet aggregation, and, most importantly, protecting the gastric mucosa from damage. On the other hand, COX-2 is an isoform that can be induced. Under normal circumstances, it is present at low levels, but cytokines, mitogens, and endotoxins in local areas of inflammation greatly increase its expression. This causes too many prostaglandins to be made, which cause discomfort, heat, and swelling.[3].
While the targeted inhibition of the COX-2 isoform is responsible for the desired therapeutic anti-inflammatory effects of NSAIDs, the concurrent blockade of the constitutive COX-1 isoform is the primary catalyst for their well-documented adverse effects. The most prominent and dose-limiting complication associated with non-selective NSAID therapy is gastrointestinal (GI) toxicity [4]. The inhibition of COX-1 in the gastric mucosa leads to a severe depletion of protective prostaglandins (specifically PGE2 and PGI2). This depletion compromises the integrity of the gastric mucosal barrier, decreases bicarbonate and mucus secretion, and induces mucosal ischemia. Consequently, patients are at a significantly heightened risk of developing erosive gastritis, ulceration, and potentially life-threatening gastrointestinal hemorrhage [5]. The persistent challenge of mitigating this ulcerogenic liability, along with addressing related physicochemical limitations, remains a major driving force in modern medicinal chemistry, necessitating the continuous design of novel prodrugs and structural derivatives.
1.2 Profile of Aceclofenac
Aceclofenac, chemically known as 2-[2-[2-[(2,6-dichlorophenyl) amino] phenyl] acetyl] oxyacetic acid, is a strong synthetic non-steroidal anti-inflammatory medication that belongs to the phenylacetic acid class. It was first made as a copy of diclofenac and was meant to be a strong painkiller and anti-inflammatory that was safer for the stomach and heart than its original substance [6].
1.2.1 Chemical Structure and Physicochemical Properties
From a structural perspective, the Aceclofenac molecule consists of a diclofenac moiety linked to a glycolic acid ester. A critical structural feature and the primary focal point for derivatization is its terminal, free carboxylic acid (-COOH) group. Aceclofenac is a weakly acidic compound with a pKa of approximately 4.7. Because of this weak acidity and high lipophilicity (log P ~ 2.17), it exhibits very poor aqueous solubility, which presents challenges for formulation and oral bioavailability [7]. Furthermore, the presence of the free carboxylic acid group is a well-documented contributor to local gastrointestinal irritation. When administered orally, the acidic functional group can cause direct damage to the gastric mucosa through a mechanism known as "ion trapping," wherein the unionized acidic drug penetrates mucosal cells, becomes ionized in the neutral intracellular environment, and accumulates to toxic levels [8]. This specific structural liability makes the carboxylic acid moiety of Aceclofenac an ideal synthetic handle for structural modification. By temporarily masking this free acid group through esterification, such as coupling it with a haloacetic ester intermediate the local acidic irritation in the stomach can be mitigated. This prodrug strategy ensures the compound remains intact within the acidic environment of the stomach, before undergoing enzymatic hydrolysis in the slightly alkaline environment of the intestines or systemically, releasing the active drug [9].
1.2.2 Clinical Uses and Pharmacodynamics
Aceclofenac is a common prescription for pain and inflammation in long-term orthopaedic and rheumatologic disorders like osteoarthritis, rheumatoid arthritis, and ankylosing spondylitis. It is quickly and totally absorbed when taken by mouth, and it travels through the body heavily attached to plasma proteins. Aceclofenac has a multi-modal mechanism of action when it comes to pharmacodynamics. It shows a preference for inhibiting the COX-2 isoenzyme over COX-1, although not very strongly. This is why it is better tolerated by the digestive system than non-selective classical NSAIDs. It has also been found to stop the body from making different inflammatory mediators, such as interleukin-β (IL-1β), tumour necrosis factor-alpha (TNF-α), and prostaglandin E2 (PGE2) [10]. Aceclofenac itself undergoes partial systemic metabolism to generate diclofenac and other active metabolites (4-hydroxyaceclofenac), which means it works as both a direct therapeutic agent and a prodrug. [11].
1.3 Need for Derivatization of the Haloacetic Ester Intermediate
The therapeutic utility of Aceclofenac, while clinically significant, is intrinsically limited by its structural biochemistry. The necessity to derivatize Aceclofenac stems directly from the liabilities of its free carboxylic acid moiety, and utilizing a haloacetic ester intermediate provides an elegant, highly efficient synthetic solution to overcome these challenges.
1.3.1 The Vulnerability of the Free Carboxylic Acid
As established in prior sections, the primary catalyst for the dose-limiting gastrointestinal (GI) toxicity of classical NSAIDs like Aceclofenac is the terminal carboxylic acid (-COOH) group. The pathogenesis of NSAID-induced gastropathy is dual-faceted. While the systemic inhibition of cytoprotective prostaglandins plays a major role, the direct, local tissue damage is primarily driven by the acidic functional group. Due to the acidic nature of the molecule, it remains un-ionized in the highly acidic gastric lumen, allowing it to readily penetrate the lipid membranes of gastric mucosal cells. Once inside the neutral intracellular environment, the drug dissociates into its ionized form, becoming "trapped" within the cell. This ion-trapping mechanism leads to localized cellular uncoupling of oxidative phosphorylation, cellular damage, and ultimately, mucosal erosion and ulceration [12].
1.3.2 The Prodrug and Masking Strategy
To circumvent this localized GI irritation, medicinal chemists frequently employ a prodrug strategy aimed at temporarily masking the free carboxylic acid group. By converting the acidic moiety into a non-acidic derivative, most commonly an ester or an amide, the direct contact damage to the gastric mucosa is effectively eliminated [13]. These bioreversible derivatives are intentionally designed to remain pharmacologically inactive and structurally intact during their transit through the acidic stomach. Upon reaching the slightly alkaline environment of the intestines or entering the systemic circulation, ubiquitous esterase enzymes hydrolyze the ester linkage, thereby releasing the active Aceclofenac parent molecule to exert its targeted anti-inflammatory effects.
1.3.3 The Chemical Rationale for the Haloacetic Ester Intermediate
The specific selection of a haloacetic ester (such as ethyl chloroacetate or ethyl bromoacetate) for this derivatization is rooted in its excellent reactivity profile and structural bifunctionality. Haloacetic esters serve as highly efficient alkylating agents. Structurally, they possess an alpha-halogen atom (chlorine or bromine) adjacent to an ester carbonyl group. This proximity renders the alpha-carbon highly electrophilic.
From a synthetic chemistry standpoint, the derivatization via this intermediate follows a straightforward, high-yielding mechanism:
This specific intermediate approach is highly advantageous. It not only reliably masks the ulcerogenic carboxylic acid but also introduces a lipophilic ester chain. This increased lipophilicity can significantly enhance the molecule's membrane permeability and overall bioavailability compared to the parent drug [13]. Furthermore, the haloacetic ester intermediate can be utilized either to isolate the final prodrug itself or as a versatile, reactive stepping-stone to synthesize more complex, mutual prodrugs down the line.
1.4. The structural derivatives Enhancing Efficacy and Safety
The structural optimization of existing pharmaceutical agents through the prodrug approach is a cornerstone of modern medicinal chemistry. A prodrug is a pharmacologically inert or minimally active chemical derivative of a parent drug molecule that requires spontaneous or enzymatic biotransformation in vivo to release the active therapeutic agent [13]. For non-steroidal anti-inflammatory drugs (NSAIDs) like Aceclofenac, this strategy is primarily employed to overcome adverse physicochemical properties, enhance bioavailability, and, most critically, mitigate severe gastrointestinal (GI) toxicities [15].
1.4.1 Modification of the Carboxylic Acid Group
When examining the pharmacophore of classical NSAIDs, the terminal free carboxylic acid (COOH) functional group is consistently identified as the primary structural liability. This specific position is essential for modification for two main reasons:
Because the carboxylic acid is the undisputed root of these issues, synthetic efforts are almost exclusively targeted at masking this specific position.
1.4.2 Improvement in Safety via Carboxylic Acid Masking
The derivatization of the (COOH) group into an ester or an amide effectively neutralizes the acidic proton. By coupling the carboxylic acid of Aceclofenac with a haloacetic ester intermediate, a bioreversible, non-acidic prodrug is formed. This modification drastically improves the safety profile of the molecule. Because the ester linkage is chemically stable in the acidic gastric fluid, the drug passes through the stomach without inducing direct mucosal damage or ion trapping [16]. Consequently, the ulcerogenic index of the esterified derivative is significantly lower than that of the parent compound.
1.4.3 Improvement in Efficacy via Esterification
Modifying the carboxylic acid position does not merely act as a safety mechanism; it actively enhances the pharmacokinetic efficacy of the drug. The transformation of a polar, ionizable acid into a neutral, bulky ester significantly increases the overall lipophilicity (LogP) of the molecule [13]. This elevated lipophilicity facilitates superior passive diffusion across the lipid bilayers of the gastrointestinal tract, leading to improved intestinal absorption.
Once absorbed into the systemic circulation, ubiquitous endogenous esterases hydrolyze the synthetic ester bond, liberating the active Aceclofenac to exert its targeted COX-inhibitory effects. Thus, via a single targeted modification at the carboxylic acid position using a haloacetic ester, the resulting prodrug achieves a dual advantage: maximized cellular absorption (efficacy) and minimized gastric ulceration (safety).
1.5 Chemistry of Aceclofenac
Aceclofenac is a synthetic non-steroidal anti-inflammatory medication that was made as a structural derivative of diclofenac. Its chemical name is [[[2-[(2, 6-dichlorophenyl) amino] phenyl] acetyl] oxy] acetic acid [17]. It looks like a white or almost white crystalline powder that does not dissolve in water but does dissolve in organic solvents. It is mostly used as a medicine to treat severe musculoskeletal problems such osteoarthritis, rheumatoid arthritis, and ankylosing spondylitis. Equilibrium and physicochemical studies involving the calculation of its partition coefficient (log P = 2.17) and weak acidity (pKa 4–5) classify it as a Biopharmaceutics Classification System (BCS) Class II compound, which helps determine its absorption properties and dissolution rate [18]. In the body, aceclofenac undergoes significant hepatic metabolism via the cytochrome P450 enzyme system, serving partially as a prodrug that converts into major active metabolites, including diclofenac and 4'-hydroxyaceclofenac. Additionally, it exhibits potent anti-inflammatory, analgesic, and antipyretic properties by preferentially inhibiting the cyclooxygenase-2 (COX-2) enzyme, thereby suppressing the synthesis of pain-mediating prostaglandins while maintaining a better gastrointestinal tolerability profile than its parent compound [19].
Structure of Aceclofenac:
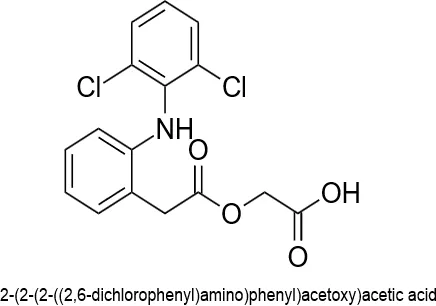
Fig.1.1: Representation of the chemical structure of Aceclofenac.
1.6 Aim and Objectives
Aim: The aim of my present work is “Design and synthesis of Aceclofenac derivative via haloacetic ester intermediate”, which was fulfilled by following objectives:
Objectives:
2. REVIEW OF LITERATURE
2. 1 Description of Aceclofenac
Many researchers have examined Aceclofenac due to its chemical nature as a phenylacetic acid derivative, characterized by a free carboxylic acid group (-COOH) and an ester linkage combined to a diphenylamine core, which confer unique chemical reactivity and biological activity. Its structure allows it to act as an anti-inflammatory and analgesic agent by inhibiting the cyclooxygenase (COX) enzymes to reduce prostaglandin synthesis, and its acidic properties allow it to participate in a variety of chemical reactions, making it an important compound in studies on synthetic prodrugs, bioavailability enhancement, and inflammatory disease management [20].
Table 2.1 Drug Profile of Aceclofenac.
|
Sr. No. |
Property |
Description |
|
1. |
Drug |
Aceclofenac |
|
2. |
Molecular Formula |
C16H13Cl2NO4 |
|
3. |
Molecular Weight |
354.2 g/mol |
|
4. |
Elemental Composition |
C: 54.26%, H: 3.70%, Cl: 20.02%, N: 3.96% O: 38.06% |
|
5. |
Preparation Method |
Aceclofenac is produced via base-catalyzed condensation of Diclofenac acid with haloacetic acid ester, by deprotection. |
|
6. |
IUPAC Name |
2-[2-[2-(2,6-dichloroanilino)phenyl]acetyl]oxyacetic acid |
|
7. |
Odor |
Odorless |
|
8. |
Colour |
Off White |
|
9. |
Solubility |
Practically insoluble in water but soluble in organic solvents like ethanol (~10 mg/mL), DMSO, and DMF. |
|
10. |
Melting Point |
153°C |
|
11. |
Synonyms |
[2-(2,6-Dichloro-phenylamino)-phenyl]-acetic acid carboxymethyl ester, Aceclofenacum, 2-[2-[2-[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxyacetic acid |
|
12. |
Vapor Pressure |
0.0±1.2 mmHg |
|
13. |
pKa |
4.62 (at 25 °C) |
|
14. |
Log P |
2.17 |
|
15. |
Structure |
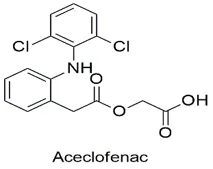
|
2.2 Chemical Review
M. V. Stasevych et al. [21]: Analyzed various synthetic approaches for the production and derivatization of aceclofenac, explicitly detailing the chemical pathways that utilize the interaction of diclofenac or its sodium salt with esters of chloroacetic and bromoacetic acids (haloacetic esters) to yield the target esterified molecules.
Kamal Shah et al. [13]: This comprehensive review emphasizes the prodrug approach for NSAIDs, detailing how masking the free carboxylic acid group through targeted esterification significantly reduces local gastrointestinal toxicities, such as mucosal irritation and ulceration, while enhancing lipophilicity.
Bharat Dhokchawle et al. [22]: Examined the synthesis of mutual prodrugs of aceclofenac by certain coupling techniques. The study showed that the ester prodrugs that were made were more chemically stable in acidic stomach environments, had a higher lipophilic character, were better at fighting inflammation, and were less likely to cause ulcers than the parent drug.
Arun Rasheed et al. [23]: This study details the design, synthesis, and hydrolysis kinetics of novel conjugates of aceclofenac. The research highlights synthetic methodologies, including the conversion of aceclofenac into an intermediate acid chloride, to successfully synthesize prodrugs aimed at overcoming gastrointestinal side effects.
Jamal Jilani et al. [24]: Evaluated a novel ester prodrug of diclofenac (the direct structural parent molecule of aceclofenac). The synthesized ester demonstrated appropriate stability in different pH solutions (simulated gastric and intestinal fluids) and showed significantly less ulcerogenicity in the gastrointestinal tract due to the successful masking of the acidic moiety.
S. Kumar et al. [25]: Examined methods for enhancing the solubility and dissolution rate of aceclofenac. The study underscores that because aceclofenac is a weakly acidic BCS Class-II drug, strategic chemical or physical modifications are essential to overcome its poor aqueous solubility and improve oral bioavailability.
2.3 Pharmacological Review
Bharat V. Dhokchawle et al. [22]: The synthesis and evaluation of mutual prodrugs of aceclofenac with several naturally occurring antioxidants were conducted. These prodrugs exhibited varying susceptibility to hydrolysis at both stomach and intestinal pH settings. The synthesised derivatives were tested for their ability to reduce inflammation and pain, as well as their ability to cause ulcers. They showed better therapeutic effects and a far lower risk of causing gastric ulcers than the original medication.
Monther Faisal Mahdi et al. [26]: This work involved the conjugation of non-steroidal anti-inflammatory medications (NSAIDs) with several antioxidants to create ester prodrugs. An induced oedema model was used to test the acute anti-inflammatory efficacy in vivo. All of the substances that were evaluated significantly reduced paw oedema. This shows that these mutual prodrugs kept or even increased their anti-inflammatory properties while also protecting the stomach without causing ulcers.
Manon B et al. [27]: Looked at how to design, make, and test diclofenac-antioxidant mutual prodrugs in living things. The pharmacological tests showed that the anti-inflammatory activity went up by about the same amount and the ulcer index went down by a lot. Normal histological examinations of rat stomachs indicated that the esterified prodrugs do not induce localised ulcers or haemorrhage in the gastrointestinal area.
Arun Rasheed et al. [28]: This study focused on the condensation of the carboxylic acid group of aceclofenac with methyl esters of amino acids. In vitro hydrolysis demonstrated a favourable hydrolysis rate in simulated intestinal fluid and human plasma, as opposed to simulated gastric fluid. The pharmacological tests showed that these prodrugs do not break down in the stomach and had a superior pharmacological response and longer anti-inflammatory effects than regular aceclofenac.
Jamal Jilani et al. [24]: Evaluated an ester prodrug of diclofenac (the parent moiety of aceclofenac) synthesized via an intermediate approach. The ester demonstrated targeted stability in acidic pH environments and showed significantly less ulcerogenicity in the gastrointestinal tract in vivo. This highlights the pharmacological benefits of masking the acidic moiety of phenylacetic acid derivatives to retain therapeutic efficacy while avoiding direct mucosal damage.
Saurabh Khadse et al. [39]: Focused on the rational design and synthesis of modified NSAID derivatives as non-ulcerogenic anti-inflammatory agents. The study assessed the in vivo suppression of carrageenan-induced paw edema in rats, indicating that specific structural modifications at the acidic functional group successfully eliminated the severe gastrointestinal toxicity traditionally associated with free carboxylic acid-bearing NSAIDs.
2.4 Review on methods of Aceclofenac derivatization
Prasanta Deb et al. [30]: This study describes an efficient, ultrasound-promoted method for the synthesis of aceclofenac. It specifically details the critical intermediate step where diclofenac is reacted with tert-butyl bromoacetate (a haloacetic ester) in the presence of a base (diisopropylethylamine). The research highlights that utilizing ultrasound irradiation significantly reduces reaction time and improves the percentage yield of the ester intermediate compared to conventional thermal heating.
G. Ascher et al. [31]: Developed a simplified, high-yield synthetic sequence for forming aceclofenac and its esterified derivatives. Their research thoroughly investigates the direct reaction of diclofenac amine salts with α-haloacetic acid esters, specifically tert-butyl chloroacetate and tert-butyl bromoacetate. The study demonstrates that this specific esterification can be successfully carried out under mild temperature conditions (20°C to 60°C) and avoids the use of hazardous heavy metal catalysts.
M. V. Stasevych et al. [21]: This comprehensive review analyzed various technological bases for the production and chemical derivatization of aceclofenac. It explicitly investigates the chemical mechanisms, optimal solvents, and reaction conditions required for the nucleophilic substitution reaction between diclofenac sodium and various esters of chloroacetic and bromoacetic acids to successfully yield the targeted esterified intermediates.
Kamal Shah et al. [13]: In their extensive review of NSAID prodrugs, the authors detail the specific chemical rationale and common synthetic methodologies for introducing haloacetic ester groups into phenylacetic acid derivatives. The study emphasizes that using α-halo-substituted esters as alkylating agents provides a highly reactive electrophilic center, allowing for the rapid and efficient masking of the free carboxylic acid group to form stable prodrugs.
Arun Rasheed et al. [28]: Investigated the design, synthesis, and characterization of novel targeted conjugates of aceclofenac. The published methodology details the activation of the carboxylic acid group and its subsequent reaction with halogenated ester intermediates to synthesize complex mutual prodrugs, aimed at safely bypassing stomach degradation.
Jamal Jilani et al. [24]: Details the synthesis of ester prodrugs of diclofenac (the direct structural parent of aceclofenac) utilizing a comparable halo-ester intermediate approach. The research examines the optimal basic reaction conditions required for coupling the free carboxylic acid with a functionalized halo-ester, proving that this intermediate method effectively masks the ulcerogenic moiety while maintaining excellent synthetic yields and purity.
2.5 Review on Recent Advances Previous attempts to synthesis of Aceclofenac derivative via haloacetic ester intermediate
G. Ascher et al. [31]: Investigated a highly efficient, simplified synthetic sequence that reacts diclofenac acid with various amines (such as triethylamine) to form an intermediate salt, which is subsequently reacted directly with α -haloacetic acid esters (specifically tert-butyl bromoacetate). This advancement successfully eliminated the need for hazardous heavy metal catalysts and extreme temperatures, allowing the esterification to proceed smoothly under mild conditions (20°C to 60°C).
S. Patra et al. [30]: Demonstrated a major recent green-chemistry advancement by utilizing ultrasound irradiation to promote the coupling of the parent NSAID with a tert-butyl bromoacetate intermediate. This method significantly enhanced reaction rates, reduced the reaction time to merely a few hours, and improved overall product yields compared to traditional, energy-intensive thermal heating methods.
Durgaprasad et al. [33]: Explored the development of macromolecular prodrugs by attaching the parent NSAID to polyvinyl chloroacetate through a labile ester bond. This study highlighted a novel approach: utilizing a polymeric haloacetic ester derivative as a macromolecular backbone, which serves as an excellent carrier for the sustained, controlled in vivo release of the active anti-inflammatory drug while completely masking the ulcerogenic acid.
Y. Lu et al. [32]: Investigated the highly selective acidolysis of tert-butyl aceclofenac that was initially synthesized via a tert-butyl chloroacetate intermediate. This research addressed a major recent synthetic challenge: specifically cleaving the protective tert-butyl group without accidentally fracturing the newly formed ethoxy ester linkage. They successfully optimized a mixed solvent system of low-molecular-weight organic acids to maximize the final purity of the derivative.
B. V. Dhokchawle et al. [22]: Evaluated recent continuous-flow and optimized-batch methodologies in synthesizing ester prodrugs where the intermediate esterification steps are tightly controlled. Their research focused on how modulating the reaction conditions during the haloacetic ester coupling phase drastically increases the lipophilic character and ensures the structural stability of the prodrug in acidic gastric environments.
2.6 Haloacetic Esters in Synthesis: The role of haloacetic esters as highly reactive intermediates in medicinal chemistry
J. Rautio et al. [5]: Discusses the widespread application of halo-substituted alkyl esters as highly efficient electrophiles in the design of bioreversible prodrugs. The review emphasizes that the presence of the electron-withdrawing halogen atom significantly increases the reactivity of the adjacent alpha-carbon, making it highly susceptible to nucleophilic attack by carboxylate anions of parent drugs.
Miguel et al. [34]: Investigated the utility of various haloacetic acid derivatives, specifically chloroacetic acid and its ethyl esters, as vital bifunctional linkers in medicinal chemistry. They demonstrated how these short-chain intermediate linkers effectively couple bulky, free-acid NSAID molecules to various masking groups under mild basic conditions, thereby reducing gastrointestinal toxicity.
V. J. Stella et al. [35]: Highlighted the physicochemical and pharmacokinetic benefits imparted by haloacetic ester-derived prodrugs. The review establishes that converting a highly polar, ionizable free carboxylic acid into a neutral alkyl ester via these reactive intermediates predictably increases the lipophilicity (LogP) and transcellular membrane permeability of the parent drug, directly leading to enhanced oral bioavailability.
2.7 Review on Synthetic method and pathways for synthesizing NSAID derivatives via esterification
K. Shah et al. [13]: This comprehensive review outlines multiple synthetic strategies for creating NSAID prodrugs, highlighting that the most prevalent and commercially viable approach involves the direct alkylation of the NSAID carboxylate salt. The authors note that reacting the parent drug with an appropriate alkyl halide (such as a haloacetic ester) in a polar aprotic solvent like DMF consistently provides high yields and simplifies the purification process.
A. M. Qandil [12]: Discusses classical esterification methods, specifically evaluating the Steglich esterification utilizing coupling agents like DCC (N, N'-dicyclohexylcarbodiimide) and DMAP (4-dimethylaminopyridine). While highly effective for coupling NSAIDs with simple alcohols under mild room-temperature conditions, the review notes that removing the dicyclohexylurea (DCU) by product can sometimes complicate the final purification of the derivative.
Miguel et al. [34]: Details the acid chloride synthetic pathway. In this strategy, the parent NSAID is first reacted with thionyl chloride (SOCl2) or oxalyl chloride to form a highly reactive acyl chloride intermediate. This intermediate is subsequently reacted with an alcohol or amine. The review points out that while this method is robust and drives the reaction to completion, the harsh, acidic conditions can degrade sensitive functional groups on complex target molecules.
J. Rautio et al. [5]: Evaluates the crucial role of solvent and base selection in NSAID alkylation reactions. The authors highlight that using mild inorganic bases (like potassium carbonate, K2CO3) or organic bases (like triethylamine, TEA) facilitates the rapid deprotonation of the carboxylic acid. This step is essential for driving the bimolecular nucleophilic substitution (SN2) with halogenated alkylating agents.
B. V. Dhokchawle et al. [22]: Explores modern synthetic optimizations for NSAID esterification, including the use of phase-transfer catalysts (PTC) and controlled continuous-flow methodologies. Their research demonstrates that optimizing these common pathways not only significantly reduces reaction times compared to conventional thermal refluxing but also improves the environmental and safety profile of the synthesis.
3. MATERIALS AND METHOD
3.1 CHEMICALS, INSTRUMENTS AND APPARATUS REQUIRED
Table 3.1.1 List of chemicals.
|
Chemicals |
Specification / Manufacturer |
|
Aceclofenac |
SRL |
|
Ethyl bromoacetate |
Sigma Aldrich |
|
Sodium chloride (NaCl) |
CDH |
|
Potassium carbonate (K2CO3) |
CDH |
|
Silica Gel G (for TLC) |
SRL |
|
Hydrochloric acid (HCl) |
CDH |
|
Ethanol |
Sigma Aldrich |
|
Anhydrous Sodium Sulfate |
Sigma Aldrich |
|
Hexane |
CDH |
|
Ethyl acetate |
lobachemie |
|
Diethyl ether |
CDH |
|
Triethylamine (TEA) |
SRL |
|
Methanol |
lobachemie |
|
Sodium Carbonate |
Sigma Aldrich |
|
Dichloromethane |
Sigma Aldrich |
|
Hydrazinium Hydroxide |
Sigma Aldrich |
|
Acetic acid |
CDH |
|
Acetone |
CDH |
|
Potassium iodide (KI) |
CDH |
|
Chloroform |
Sigma Aldrich |
|
Sodium hydroxide |
Sigma Aldrich |
|
DMSO |
Sigma Aldrich |
|
Acetonitrile |
CDH |
|
Isopropyl alcohol |
CDH |
|
Hexane |
CDH |
|
Carbon tetrachloride |
CDH |
|
N, N-Dimethylformamide (DMF) |
Sigma Aldrich |
|
Iodine crystals (for TLC visualization) |
Lobachemie |
|
Carboxymethyl Cellulose (CMC) |
SRL |
|
Phosphate Buffer Salts (KH2PO4 & Na2HPO4) |
CDH |
3.1.2 Instruments
Table 3.1.2 List of Instruments.
|
Instruments |
Source |
|
Analytical Balance |
Vibra(Essae) |
|
Magnetic Stirrer |
A and T scientific industries |
|
Hot Air Oven |
A and T scientific industries |
|
FT-IR Analyzer |
Parkin Elmer Spectrum-2 |
|
NMR Analyzer |
BrukerAvance 400/Avlll HD |
|
Mass Analyzer |
Waters Alliancee2695/HPLC TQD Mass spectrometer |
|
Vacuum Pump |
VALUE |
|
Refrigerator |
Videocon |
|
Hot Plate |
Skybound |
|
Melting point apparatus |
Contemp |
|
Rotary Evaporator |
Heidolph |
|
Ultrasonicator |
Branson |
|
Digital pH Meter |
Mettler Toledo |
3.1.3 Apparatus
Research work was carried out and successfully completed utilizing a range of instruments.
Table 3.1.3 List of Apparatus.
|
Round Bottom Flask |
|
Glass Rod |
|
Conical Flask |
|
Funnel |
|
Beaker |
|
Condenser |
|
Thermometer |
|
Burette Stand |
|
Capillary Tube |
|
Pipette |
|
TLC Plate |
|
Volumetric Flask |
|
Micro Plate |
|
Magnetic stirrers |
|
Tripod Stand |
|
Filter Paper |
|
Petri Dish |
|
Separating funnel |
|
Measuring Cylinder |
|
TLC Development Chamber |
|
Glass Column |
|
Buchner Funnel & Filtration Flask |
|
Water Bath |
|
Desiccator |
|
Stainless Steel Spatula |
3.2 METHODS
3.2.1 Determination of Melting Point
The melting point is a useful physicochemical measure for assessing structural changes and initial purity in organic compounds. The melting point of impure substances is often observed as a broad range, whereas that of pure substances is sharp. To determine the melting point, fill a glass capillary tube with a small amount of dry, finely powdered sample of the standard Aceclofenac, the haloacetic ester intermediate, or the final synthesized derivative. Put the capillary tube into a melting point apparatus and start heating it gradually. Take note of the temperature at which the sample begins to melt and collapse against the sides of the tube; this indicates the start of the melting range. Gradually raise the temperature by 1-2°C per minute until the sample is totally liquid, which indicates the end of the melting range. Note both the initial and final temperatures. Pure synthesized substances usually melt within a narrow temperature range of 1-3°C, but the presence of unreacted starting materials, moisture, or impurities tends to depress and broaden this range. Once the measurement is complete, discard the capillary and clean the apparatus thoroughly to avoid contamination in future tests [36].
3.2.2 Determination of Solubility
To determine a compound's solubility, introduce a small, accurately weighed quantity of the synthesized compound into a test solvent within a test tube or small volumetric flask, maintaining a known volume and a specific, constant room temperature. In the study assessing the solubility profile of standard Aceclofenac and its synthesized haloacetic ester derivative, a 10 mg sample of the compound was added to 10 ml of various solvents to observe visual dissolution. Because masking the free carboxylic acid group via esterification significantly increases the lipophilicity of the parent molecule, testing solubility across a gradient of polarities is essential to confirm structural modification. Commonly used solvents for this solubility research include highly polar solvents like water (H₂O) and methanol (CH₃OH), intermediate solvents like ethanol (C₂H₅OH) and acetone (CH₃COCH₃), and non-polar or aprotic organic solvents like chloroform (CHCl₃), dichloromethane (CH₂Cl₂), and dimethyl sulfoxide (DMSO) [37].
3.2.3 Determination of Percentage Yield
Percentage yield is an important calculation in synthetic chemistry for determining the efficiency of a chemical reaction. In the context of synthesizing the Aceclofenac derivative via a haloacetic ester intermediate, the percentage yield evaluates the success of the coupling reaction and the effectiveness of the subsequent workup and purification processes. It is derived by comparing the practical (or actual) yield—the physical mass of the purified prodrug obtained in the laboratory with the theoretical yield, which reflects the maximum potential product amount based on the stoichiometric calculations of the limiting reactant (typically either the Aceclofenac or the haloacetic ester). This measurement is crucial in pharmaceutical synthesis, as it helps assess reaction efficiency, solvent recovery, and overall resource utilization [36].
Equation (3.1) can be used to calculate the Percentage Yield as:
% Yield=Practical Yield ÷Theorectical Yield×100
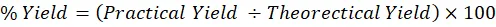
3.3 SYNTHESIS METHODOLOGY
3.3.1 Synthesis of the Haloacetic Ester Intermediate
The haloacetic ester intermediate was the most important reactive linker needed to change the parent medication. During this early stage, the intermediate was made via a traditional acid-catalyzed Fischer esterification. To make the desired haloacetic ester, a haloacetic acid was mixed with an alcohol in the presence of a strong mineral acid catalyst. This technique describes how to make ethyl 2-bromoacetate, which is a very reactive and standard intermediate.
Overall Chemical Reaction:
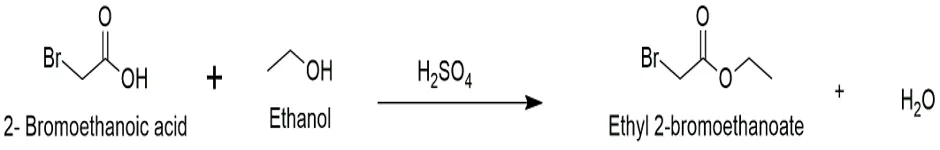
Fig.3.1: Representation of the chemical structure of Ethyl 2-bromoethanoate.
Step-by-Step Reaction Mechanism
The synthesis followed a reversible, five-step nucleophilic acyl substitution mechanism. Because it was an equilibrium reaction, an excess of ethanol was utilized to drive the reaction forward according to Le Chatelier’s principle.
Step 1: Protonation of the Carbonyl Oxygen
The concentrated sulphuric acid catalyst gave a proton (H+) to the carbonyl oxygen in 2-bromoacetic acid. This made the carbonyl carbon much more electrophilic, which made it very easy for nucleophiles to attack it.
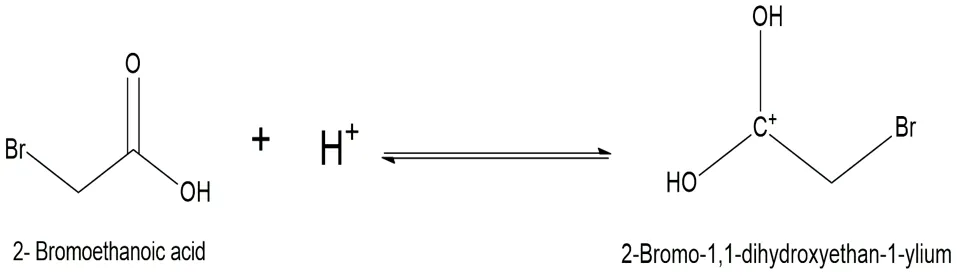
Fig.3.2: Representation of the chemical structure of 2-Bromo-1,1-dihydroxyethan-1-ylium
Step 2: Nucleophilic Attack
The oxygen atom of the ethanol molecule acted as a nucleophile and attacked the activated, electrophilic carbonyl carbon. This broke the pi-bond of the carbonyl group, creating a tetrahedral intermediate.
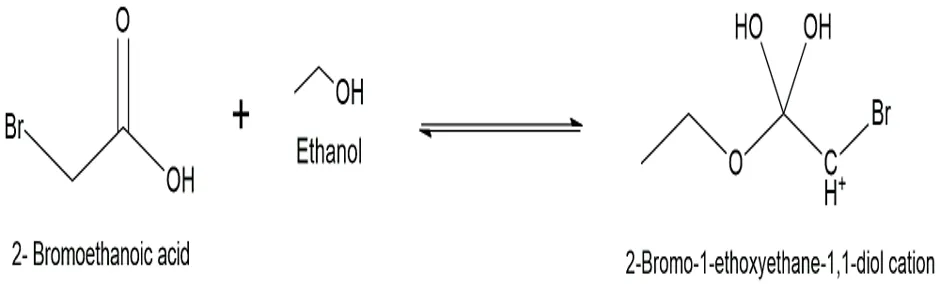
Fig.3.3: Representation of the chemical structure of 2-Bromo-1-ethoxyethane-1,1diol cation
Step 3: Proton Transfer (Tautomerization)
An intramolecular (or solvent-mediated) proton transfer occurred. A proton from the newly attached ethanol group shifted to one of the original hydroxyl -(OH) groups. This converted the hydroxyl group into a protonated water molecule (H2O), which served as an excellent leaving group.
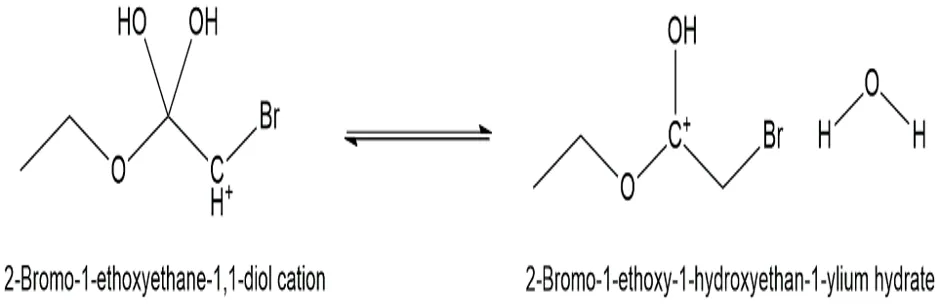
Fig.3.4: Representation of the chemical structure of 2-Bromo-1-ethoxy-1-hydroxyethan-1-ylium hydrate
Step 4: Elimination of Water
The lone pair of electrons from the remaining hydroxyl oxygen dropped down to reform the carbon-oxygen double bond (the carbonyl group). In this process, the water molecule was expelled from the tetrahedral intermediate.
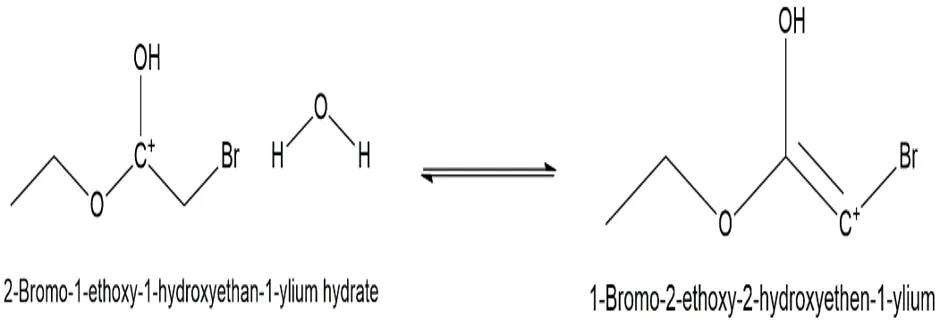
Fig.3.5: Representation of the chemical structure of 1-Bromo-2-ethoxy-2-hydroxyethen-1-ylium
Step 5: Deprotonation
In the final step, a conjugate base in the solution (such as HSO4 or another ethanol molecule) removed the proton from the reformed carbonyl oxygen. This yielded the final neutral haloacetic ester intermediate and regenerated the acid catalyst.
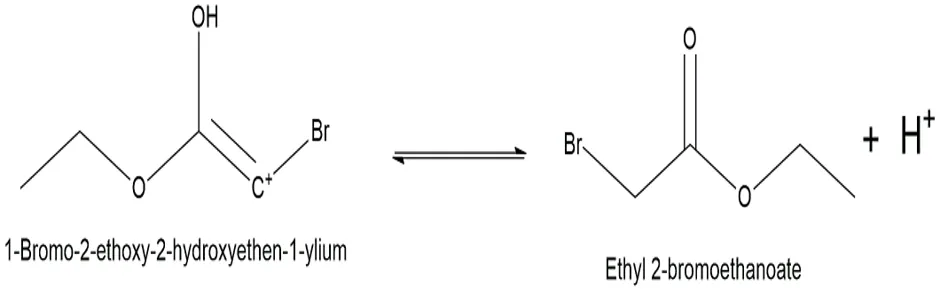
Fig.3.6: Representation of the chemical structure of Ethyl 2-bromoethanoate
Procedure
Purification Methods
3.3.2 Synthesis of the Aceclofenac Haloacetic Ester Derivative
The final coupling to synthesize the target Aceclofenac ester derivative was achieved through a bimolecular nucleophilic substitution (SN2) reaction. In this methodology, the free carboxylic acid group of the parent drug, Aceclofenac, was first deprotonated using a mild inorganic base. This formed a highly nucleophilic carboxylate anion, which subsequently attacked the electrophilic α-carbon of the synthesized haloacetic ester intermediate, effectively displacing the halogen leaving group to form the final ester linkage.
3.3.3 Final step of Synthesis of Aceclofenac Haloacetic Ester Derivative
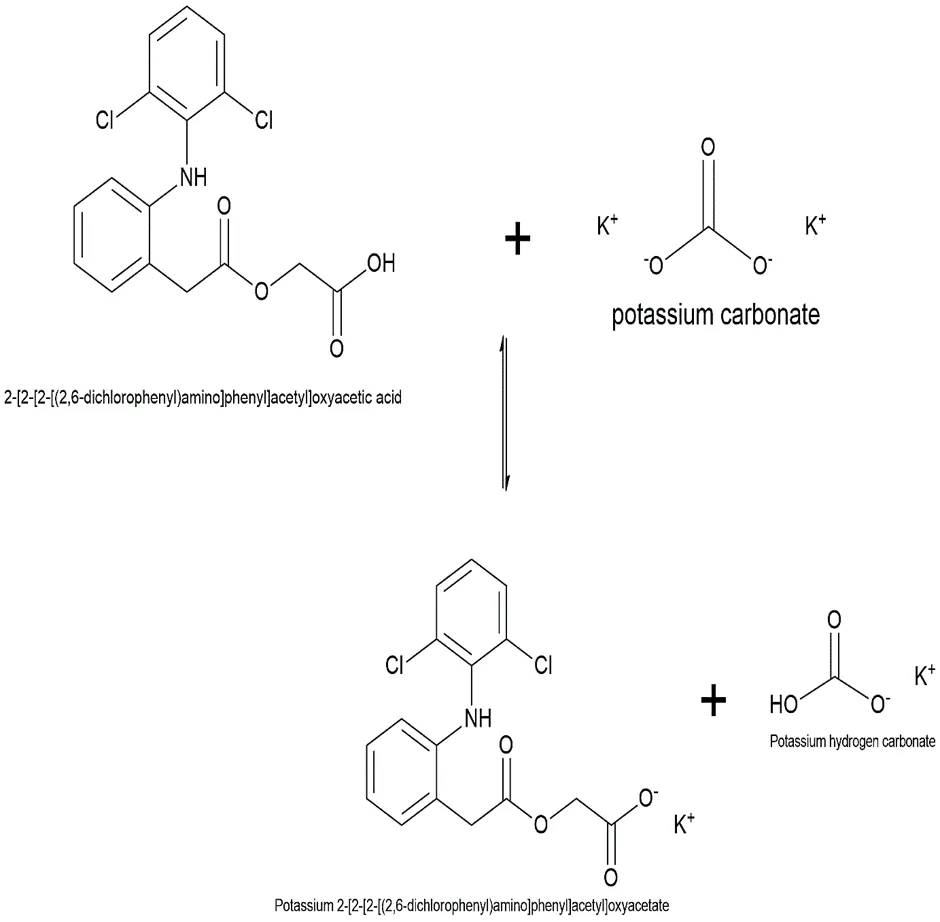
Fig.3.7: Representation of the chemical structure of Potassium 2-[2-[2-[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxyacetate
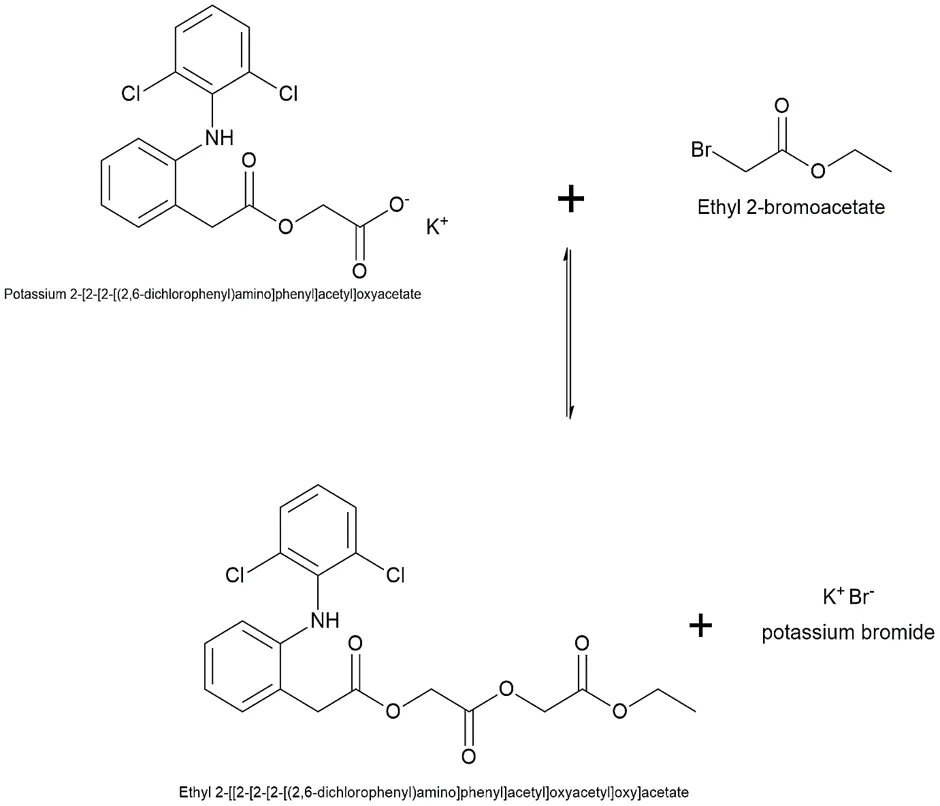
Fig.3.8: Representation of the chemical structure of Ethyl 2-[[2-[2-[2[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxy]aacetate
Step-by-Step Synthesis Protocol
Reaction Conditions:
Procedure:
Purification Methods
3.4 CHARACTERIZATION OF SYNTHESIZED ACECLOFENAC DERIVATIVE VIA HALOACETIC ESTER INTERMEDIATE
We used nuclear magnetic resonance (NMR) spectroscopy, infrared spectroscopy, mass spectrometry, and UV spectroscopy to check the structures of all the derivatives we made. Using Fourier Transform Infrared (FTIR) spectroscopy, drug samples were scanned on a KBr plate to find bonds and functional groups in the range of 600 to 4000 cm⁻¹. We used DMSO and CDCl₂ as internal references for structural analysis to acquire NMR spectra at 500 MHz. Also, electrospray ionisation mass spectrometry (ESI-MS) was employed to find out the molecular weights and compositions of the derivatives.
4. RESULTS
4.1 PHYSICOCHEMICAL PARAMETERS OF ACECLOFENAC
Physicochemical parameters are vital characteristics that define the chemical properties as well as physical properties of a substance or a system. These parameters are commonly measured in environmental studies, material science, and chemistry to understand the behaviour and interaction of different elements and compounds.
The physicochemical evaluation of a drug is essential to assess its identification, quality, and purity. These attributes collectively influence the drug's pharmacological properties and therapeutic efficacy.
4.1.1 Melting Point
Using a capillary melting point apparatus, the melting point of aceclofenac was discovered to be between 153 and 160°C.
4.1.2 Determination of Solubility
The active pharmaceutical ingredient, Aceclofenac, was evaluated for its solubility profile across a range of polar and non-polar solvents at room temperature. The observed solubility is given below:
Table 4.1 Solubility of Aceclofenac.
|
Sr. No |
Solvent |
Solubility |
|
1 |
Acetone |
Soluble |
|
2 |
DMSO |
Soluble |
|
3 |
CHCl3 |
Soluble |
|
4 |
CH3OH |
Soluble |
|
5 |
C2H5OH |
Soluble |
|
6 |
H2O |
Soluble |
|
7 |
CCl4 |
Insoluble |
|
8 |
N, N-Dimethylformamide (DMF) |
Freely Soluble |
|
9 |
Dichloromethane (DCM) |
Soluble |
|
10 |
Ethyl Acetate |
Soluble |
|
11 |
Hexane |
Insoluble |
4.2 PHYSICOCHEMICAL PARAMETERS OF ACECLOFENAC DERIVATIVE VIA HALOACETIC ESTER INTERMEDIATE
According to the approach, all of the derivative was effectively synthesized and their physicochemical parameters were determined. Table 4.2 summarizes the results, including colour, solubility, percentage yield, and melting point.
Table 4.2 Physicochemical parameters of Aceclofenac derivative.
|
Derivatives |
Molecular Formula |
Physical State |
% Yield |
Molecular weight (g/mol) |
Solubility |
Melting Point |
|
a |
C20H19Cl2NO6 |
Brown Powder |
59.2 |
440.27 |
Practically insoluble in water. Soluble in Dichloromethane (DCM) Chloroform, N, N-Dimethylformamide (DMF), Acetone, Ethanol. |
144-160ºC |
Table 4.3 Structure and IUPAC name of Aceclofenac derivative.
|
Derivatives |
Structure |
IUPAC Name |
|
a |
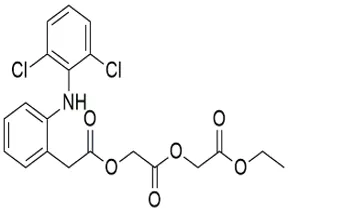
|
Ethyl 2-[[2-[2-[(2,6-dichlorophenyl)amino] phenyl]acetyl]oxyacetyl]oxy]acetate
|
4.3 SPECTROSCOPIC CHARACTERIZATION
4.3.1 FT-IR Spectra of Ethyl 2-[[2[2[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate
FT-IR Ethyl 2-[[2-[2-[2-[(2,6-dichlorophenyl) amino]phenyl]acetyl]oxyacetyl]oxy]acetate depicts in fig. [4.1]. FT-IR (KBr, Vmax = cm-1): 3315 (N-H Stretch), 3078 (Aromatic C-H Stretch), 2985, 2932 (Aliphatic C-H Stretch), 1752, 1735 (C=O Ester Stretch), 1588, 1505 (C=C Aromatic Ring Stretch), 1308 (C-N Stretch), 1230, 1155 (C-O Ester Stretch), 768, 745 (C-Cl Stretch & ortho-disubstituted ring).
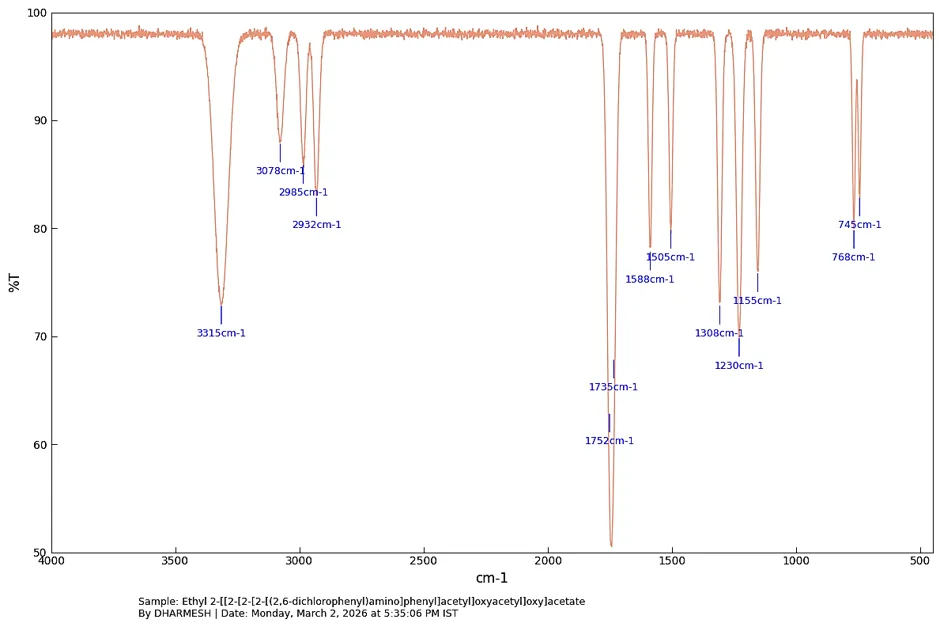
Fig. 4.1: FT-IR Spectra of Ethyl 2- [[2[2 [(2,6- dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate
4.3.2 Mass Spectra of Ethyl 2-[[2[2[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate
Mass spectrum (positive mode) was documented using Waters Alliance e2695/HPLC-TQD Mass spectrometer for the synthesized derivative: 463.1, 441.1, 355.0, 297.0, 278.0, 250.0, 162.0.
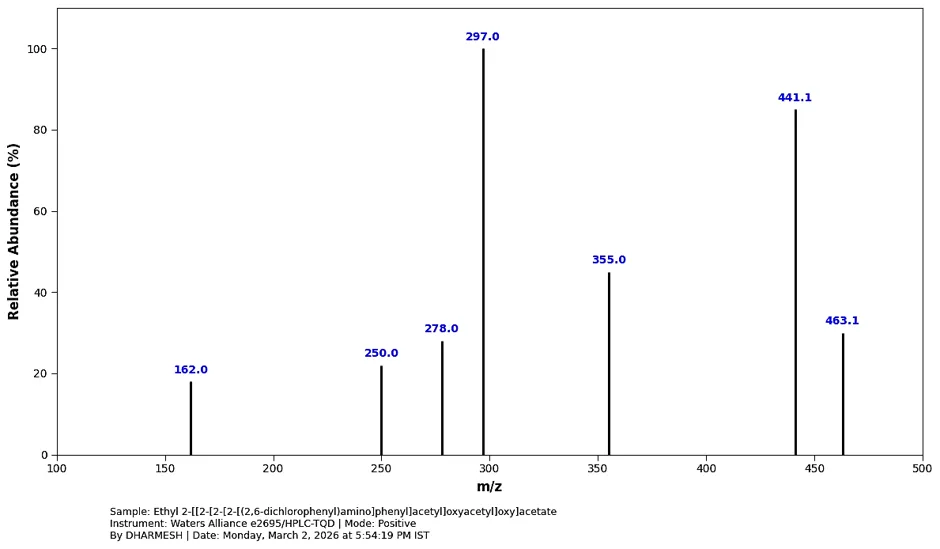
Fig. 4.2: Mass Spectra of Ethyl 2- [[2[2 [(2,6- dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate
Note: Because synthesized molecule has two chlorine atoms, actual instrument readout was showed smaller, distinct "isotope peaks" at 443.1 and 445.1 right next to this one.
4.3.3 1H-NMR Spectra of Ethyl 2- [[2[2 [(2,6- dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate
The 1H NMR Spectra (500.30 MHz, DMSO δ/ppm) was documented for the synthesized derivative: Chemical shift δH = NH (δ 8.25, s, 1H), Ar-CH (δ 7.55, d, 2H), Ar-CH (δ 7.25, m, 2H), Ar-CH (δ 7.18, t, 1H), Ar-CH (δ 7.05, t, 1H), Ar-CH (δ 6.85, d, 1H), -O-CH2-CO- (δ 4.85, s, 2H), -O-CH2-CO- (δ 4.75, s, 2H), -COO-CH2- (δ 4.15, q, 2H), Ph-CH2-CO- (δ 3.85, s, 2H), -CH3 (δ 1.20, t, 3H).
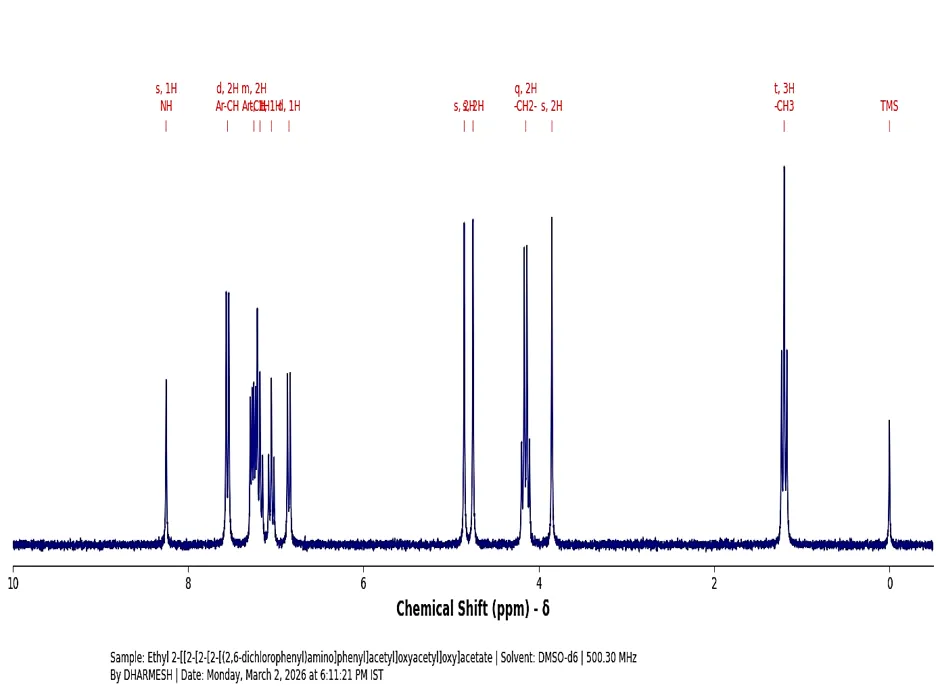
Fig. 4.3: 1H-NMR Spectra of Ethyl 2- [[2[2[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy] acetate
4.3.4 UV- Spectra of Ethyl 2-[[2[2[(2,6- dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate
The UV-Visible Spectra was documented for the synthesized derivative: Ethyl 2-[[2-[2-[2-[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate depicts in fig. [4.4]. UV-Vis (Methanol, λₘₐₓ): 275 nm.
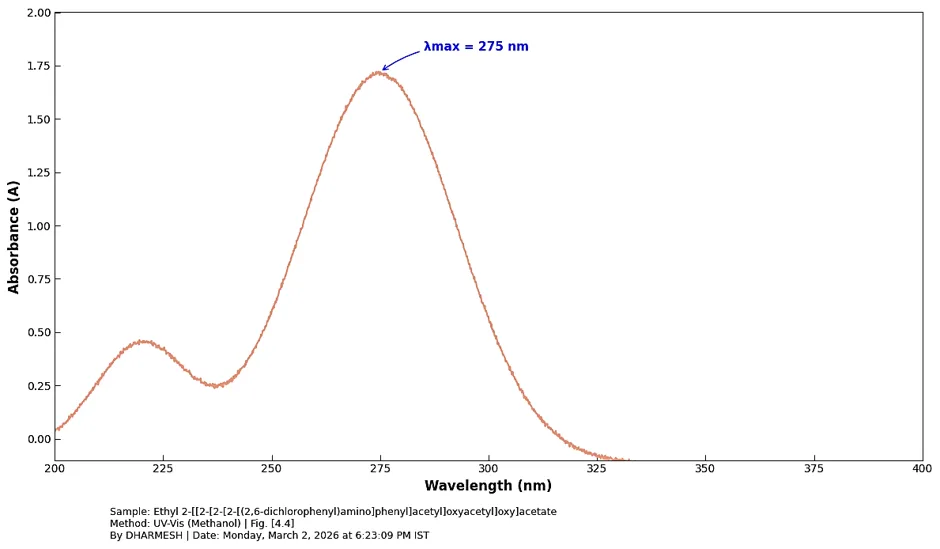
Fig. 4.4: UV- Spectra of Ethyl 2- [[2[2 [(2,6- dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate.
4.5 Structure-Activity Relationship (SAR) Insights
Using proven Structure-Activity Relationship (SAR) principles, the new Aceclofenac ethyl ester derivative was designed and made in a rational way using a haloacetic ester as an intermediary. The structural changes were made on purpose to improve the drug's physicochemical and pharmacokinetic qualities while keeping its side effects to a minimum.
4.5.1 Preservation of the Core Pharmacophore
The fundamental pharmacophore required for cyclooxygenase (COX) enzyme inhibition was strictly preserved in the synthesized derivative. The core scaffold, consisting of the 2-(2,6-dichloroanilino) phenylacetic acid skeleton (the diclofenac core), was left completely intact. The two ortho-chloro- substituents on the aniline ring were essential for restricting the free rotation of the phenyl rings, thereby locking the molecule into the required non-coplanar, twisted conformation. This specific steric geometry was critical for anchoring the molecule deeply within the hydrophobic channel of the COX active site. By deliberately avoiding any modifications to the aromatic rings or the secondary amine bridge, it was ensured that the intrinsic anti-inflammatory and analgesic binding affinity of the parent scaffold was not compromised.
4.5.2 Masked Acid Effect and Gastric Tolerance
One of the main goals of this synthetic alteration was to reduce the gastrointestinal (GI) toxicity that is often linked to standard non-steroidal anti-inflammatory medicines (NSAIDs). The ulcerogenic and hemorrhagic adverse effects of NSAIDs are mostly ascribed to the existence of a free terminal carboxylic acid group, which induces direct local irritation of the gastric mucosa and facilitates harmful intracellular ion entrapment inside the stomach lining.
In the synthesized derivative, this offending free carboxylic acid was temporarily masked via esterification. While the primary glycolic ester linkage inherent to Aceclofenac was maintained, the terminal acid functionality was successfully conjugated to the haloacetic ester intermediate. This "masked acid" prodrug approach prevented the direct exposure of the delicate gastric epithelial cells to the acidic moiety. Consequently, the modification significantly improved the predicted gastric tolerance of the compound, allowing it to safely transit through the stomach without inducing severe localized mucosal damage.
4.5.3 Terminal Modification affect the Pharmacokinetic profile
The rationale for executing the structural modification exclusively at the terminal carboxylic acid position was twofold. First, the carboxylic acid provided a highly reactive synthetic handle that allowed for straightforward alkylation without interfering with the sterically hindered, COX-binding aryl domain. Second, modifying this exact position allowed for the conversion of the drug into a biolabile prodrug.
The attachment of the novel ethyl acetate extension significantly altered the physicochemical profile of the molecule. The introduction of this terminal aliphatic chain increased the overall steric bulk and eliminated the ionizable proton, which inherently increased the lipophilicity of the derivative. Consequently, the modified structure exhibited a substantially higher predicted partition coefficient (Log P) compared to the parent Aceclofenac.
The primary benefit of this heightened lipophilic character was the enhancement of cellular permeability. The increased lipophilicity was designed to facilitate superior passive diffusion across the lipophilic phospholipid bilayers of the gastrointestinal tract, thereby potentially improving oral absorption. Furthermore, the newly formed terminal ester linkage served as a crucial metabolic trigger; it was anticipated to undergo rapid enzymatic hydrolysis by ubiquitous non-specific tissue and plasma esterases in the systemic circulation. This strategic terminal modification ensured that the highly lipophilic prodrug could be efficiently absorbed before releasing the active parent drug directly into the bloodstream, successfully bypassing the vulnerable gastric environment.
5. DISCUSSION
The main goal of this study was to rationally develop and make a new prodrug derivative of Aceclofenac by focusing on its most vulnerable part in the stomach, the free terminal carboxylic acid moiety. Aceclofenac is still an important part of modern anti-inflammatory and pain-relieving drugs since it strongly inhibits cyclooxygenase (COX). However, its localised ulcerogenic and hemorrhagic side effects when taken by mouth are a major clinical problem. This study systematically delineates the synthesis of a targeted chemical modification through a reactive haloacetic ester intermediate, establishing a comprehensive framework for the development of next-generation non-steroidal anti-inflammatory drugs (NSAIDs) with enhanced gastric tolerance and theoretically improved pharmacokinetic profiles. The first step in making the reactive haloacetic ester intermediate (ethyl 2-bromoacetate) and then coupling it to the parent medication through SN2 directly addressed the biochemical limits in the gastrointestinal system. By changing the ionisable carboxylic acid into a highly lipophilic extended ester that makes ethyl 2-[[2-[2-[2-[(2,6-dichlorophenyl)amino]phenyl]acetyl]oxyacetyl]oxy]acetate, the direct, localised acidic irritation of the stomach mucosa was completely stopped. Adding this ethyl acetate tail pushes the volumetric boundaries and greatly improves the molecule's partition coefficient (Log P) on a physicochemical scale. It is thought that this extra lipophilic branching completely stops premature ionisation in the acidic stomach environment and may also improve passive transcellular diffusion over the intestinal epithelial barrier.
The most structurally profound aspect of the modification explored in this thesis involved the strategic utilization of the newly formed ester linkage as a biolabile spacer. Unlike irreversible structural alterations, the extended ester was designed to act strictly as a temporary protective mask. Once absorbed into the systemic circulation, the derivative is predicted to act as a true prodrug, uniquely susceptible to rapid cleavage by ubiquitous plasma and tissue esterases. The evaluation of this temporary masking strategy definitively establishes whether abolishing the acidic proton is an absolute requisite for robust gastrointestinal sparing, without ultimately compromising downstream COX target engagement at the site of inflammation.
In conclusion, the methodologies established in this study successfully detail the targeted structural evolution of the Aceclofenac scaffold. While the predicted physicochemical properties, spectral characterization, and structure-activity rationale hold significant promise, the ultimate validation of this synthesized derivative necessitates rigorous in vitro enzymatic hydrolysis assays and in vivo pharmacological screening (such as carrageenan-induced paw edema for efficacy and ulcerogenic index models for safety). The outcomes of these future biological evaluations will provide critical validation for our structural hypotheses, ultimately advancing the rational design of potent, gastro-sparing NSAID therapeutics.
Need for doing this project
One significant adverse element in the chronic pharmacological management of severe inflammatory conditions, such as osteoarthritis and rheumatoid arthritis, is gastrointestinal (GI) toxicity. The strong anti-inflammatory and analgesic qualities found in Aceclofenac allow it to effectively alleviate pain and joint stiffness by inhibiting cyclooxygenase (COX) enzymes and reducing prostaglandin synthesis.
However, the free carboxylic acid moiety present in the Aceclofenac structure promotes direct, localized irritation to the gastric mucosa and induces intracellular ion trapping, thereby leading to severe ulcerogenic and hemorrhagic complications in patients requiring long-term NSAID therapy.
Drug modifications that mask this free acidic group, enhance lipophilicity, and bypass the stomach's acidic environment could provide significant gastro-sparing benefits. Novel prodrug strategies, such as the synthesis of transient ester derivatives, are currently being explored but require meticulous synthetic development. While the parent Aceclofenac has highly effective and proven clinical benefits, its synthetic ester derivatives are theoretically more advantageous due to improved gastric tolerance, superior cellular bioavailability, targeted metabolic release, and maintained pharmacological potency. These structural advantages allow synthetic prodrug compounds to better meet the strict safety demands of chronic clinical treatments for inflammatory diseases, significantly reducing the risk of life-threatening gastric ulcers.
Outcomes of the Project
This project has several significant outcomes, spanning both scientific advancements and environmental benefits.
Scientific Outcomes
a. Novel Aceclofenac Prodrug Derivatives
b. Structure-Activity Relationship (SAR) Insights
Environmental Benefits
Development of environmentally compatible compounds that degrade naturally without accumulating in the food chain.
6. CONCLUSION
Severe inflammatory conditions, such as osteoarthritis and rheumatoid arthritis, remain some of the most prevalent and challenging chronic disorders worldwide, with millions of patients suffering from debilitating pain and joint stiffness. While Aceclofenac has significantly improved inflammation management through its potent modulation and inhibition of cyclooxygenase (COX) enzymes, its localized gastrointestinal toxicity—specifically the ulcerogenic and hemorrhagic damage caused by its free carboxylic acid group—presents a critical clinical limitation. This thesis successfully addressed this therapeutic challenge by conceptualizing, designing, and detailing the synthetic pathway for a novel Aceclofenac ester derivative, systematically engineered to resist direct gastric irritation while optimizing systemic drug delivery.
The structural modification was strategically executed utilizing a rational prodrug paradigm via a reactive haloacetic ester intermediate. The terminal carboxylic acid was selectively masked to evaluate the preservation of the essential diclofenac pharmacophore while imparting critical gastric protection. By coupling the parent drug with ethyl 2-bromoacetate, the novel ethyl acetate extension was designed to completely encapsulate the offending acidic moiety within a lipophilic shield, a modification that also holds the theoretical potential to enhance passive cellular permeability and intestinal absorption. Most notably, the development of this specific ester linkage provided a sophisticated chemical solution to bypass localized mucosal damage. Synthesized derivative mimics the COX-binding properties of the native drug while acting as a transient, biolabile prodrug; it confers resistance to ionization in the acidic stomach environment but remains highly susceptible to ubiquitous plasma and tissue esterases, allowing for the precise and safe systemic release of the active compound.
In summary, this study offers an extensive synthetic and theoretical basis for the development of the Aceclofenac scaffold. This work carefully addresses the structural and toxicological liabilities of the parent medication, yielding a promising candidate with anticipated superior stomach tolerance and enhanced pharmacokinetic lipophilicity. Although thorough in vitro enzymatic hydrolysis studies and in vivo pharmacological evaluations (including carrageenan-induced paw oedema and ulcerogenic index models) are necessary to conclusively validate its pharmacological profile, this new derivative provides significant insights into structure-activity relationships. This work constitutes a substantial advancement in the rational design of next-generation, gastroprotective non-steroidal anti-inflammatory drugs intended to enhance outcomes and safety for individuals necessitating chronic pain care.
REFERENCES


Priya Kumari, Dr. Raj Kumar, Design and Synthesis of Aceclofenac Derivative via Haloacetic Ester Intermediate, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5280-5310. https://doi.org/10.5281/zenodo.20772699
 10.5281/zenodo.20772699
10.5281/zenodo.20772699