
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
S. N. College of Pharmacy
Acid-related gastrointestinal disorders, including Gastro-oesophageal Reflux Disease (GERD) and Peptic Ulcer Disease (PUD), constitute a substantial global healthcare burden. Proton Pump Inhibitors (PPIs), such as Pantoprazole, are the most important drugs for treating these diseases. They work by covalently and permanently blocking the stomach H+/K+ ATPase enzyme. Despite its clinical efficacy and different pH-dependent activation profile, Pantoprazole exhibits significant pharmacokinetic limitations, primarily its short plasma elimination half-life and high susceptibility to hepatic CYP2C19-mediated metabolism. These limitations frequently result in nocturnal acid breakthrough and require repeated, larger dosing. The metabolic weakness of Pantoprazole is primarily localized at its central sulfoxide (S=O) pharmacophore. To address these critical therapeutic gaps, this research aimed to design and synthesize a novel Sulfoxamide Modified Pantoprazole Analogue. The core strategy involved the bioisosteric replacement of the metabolically labile sulfoxide oxygen with a nitrogen species to form a sulfoxamide core (S(O)=NH). This rational modification was hypothesized to enhance metabolic resistance against CYP450 enzymes, extend the drug's plasma half-life, and maintain the essential tetrahedral geometry necessary for target affinity at the proton pump, while simultaneously reducing the environmental accumulation of active pharmaceutical ingredients through the promotion of targeted, eco-friendly catalytic synthesis. The methodology encompassed a rational three-step synthetic pathway. Step 1 involved the synthesis of the acid-stable scaffold, 5-(difluoromethoxy)-2-mercaptobenzimidazole (Derivative 3a). In Step 2, this core was coupled with 2-chloromethyl-3,4-dimethoxypyridine to yield the thioether intermediate, Pro-Pantoprazole sulfide (Derivative 3b). The final and pivotal Step 3 utilized a novel metal-catalyzed oxidative imination (sulfoximination) employing Rhodium (II) acetate dimer as a catalyst, ammonium carbamate as the nitrogen source, and (Diacetoxyiodo)benzene (PIDA) as the oxidant to successfully yield the targetsulfoxamide analogue (Derivative 3c). The synthesized derivatives (3a, 3b, and 3c) were isolated, purified using column chromatography and recrystallization, and subjected to rigorous physicochemical and spectroscopic characterization. The percentage yields for the compounds demonstrated efficient synthesis, recorded at 55%, 73.5%, and 83.4%, respectively. Structural confirmation was definitively achieved through Fourier-Transform Infrared (FT-IR) spectroscopy, Proton Nuclear Magnetic Resonance (1H-NMR), and Mass Spectrometry. Crucially, the FT-IR spectra of the final target compound (3c) revealed characteristic N-H (3277.32cm-1) and 3123.59cm-1 stretching vibrations, while 1H-NMR and mass spectra (m/z 412.2) conclusively validated the successful integration of the sulfoxamide moiety into the Pantoprazole scaffold.
1.1 Background on Peptic Ulcers and Gastroesophageal Reflux Disease (GERD)
Acid-related disorders represent a significant category of gastrointestinal pathologies that affect a vast proportion of the global population, contributing substantially to morbidity and healthcare costs [Engevik et al., 2020]. These problems mostly happen when there is an imbalance between hostile substances like gastric acid (hydrochloric acid) and pepsin and the body's defences that keep the mucosa healthy, like bicarbonate secretion, mucus production, and good blood flow [Laine et al., 2016]. When the physiological regulation of gastric acid secretion is disrupted, or when mucosal defenses are overwhelmed, it leads to a spectrum of conditions ranging from mild dyspepsia to severe ulcerative diseases.
Gastro-oesophageal Reflux Disease (GERD) is likely the most common of these disorders. It is characterised as a chronic condition in which the retrograde movement of gastroduodenal contents into the oesophagus results in distressing symptoms and possible mucosal damage [Vakil et al., 2006]. The pathogenesis of GERD is multifaceted, primarily influenced by the transitory relaxation of the Lower Esophageal Sphincter (LES), compromised esophageal clearance, and delayed stomach emptying. The chronic exposure of the esophageal epithelium to acidic refluxate can lead to erosive esophagitis and, in severe cases, Barrett’s esophagus, which is a precursor lesion for esophageal adenocarcinoma [Maregalia et al., 2012]. The condition is clinically characterized by persistent heartburn and acid regurgitation, significantly impairing the patient's quality of life.
Peptic Ulcer Disease (PUD) persists as a significant clinical problem, marked by the development of discrete lesions in the mucosal lining of the stomach (gastric ulcer) or the proximal duodenum (duodenal ulcer), penetrating the muscularis mucosae [Lanas & Chan, 2017]. The discovery of Helicobacter pylori (H. pylori), a gram-negative bacterium that colonises the stomach antrum and damages mucosal protective barriers, has transformed the understanding of the aetiology of PUD. In addition to infectious reasons, the prevalent utilisation of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) serves as a significant secondary factor. NSAIDs block cyclooxygenase (COX) enzymes, which lowers the production of cytoprotective prostaglandins and makes the mucosa more likely to be damaged by acid [Malfertheiner et al., 2009].
Gastric acid hypersecretion is a defining characteristic of Zollinger-Ellison Syndrome (ZES), in addition to these other illnesses. Zollinger-Ellison syndrome (ZES) is a serious disorder caused by a gastrin-secreting neuroendocrine tumour (gastrinoma) that is usually found in the pancreas or duodenum. The tumor releases excessive amounts of gastrin, stimulating the parietal cells to secrete massive volumes of hydrochloric acid, leading to intractable peptic ulcers and diarrhea [Jensen et al., 2021].
Despite the distinct etiologies of GERD, PUD, and ZES, they share a common therapeutic target: the reduction of gastric acidity. This shared pathophysiology underscores the critical importance of pharmacological agents capable of effectively suppressing acid secretion, specifically Proton Pump Inhibitors (PPIs), which target the final pathway of acid production—the H+/K+ ATPase enzyme [Shin & Kim, 2013].
The gastric H+/K+ ATPase enzyme, sometimes called the "proton pump," is what makes gastric acid secretion happen. This enzyme is a member of the P-type ATPase family and is only found in the apical secretory membranes of stomach parietal cells [Shin et al., 2009]. This enzyme's job in the body is to use the energy from breaking down ATP to speed up the electroneutral exchange of cytoplasmic hydrogen ions (H+) for extracellular potassium ions (K+). This active transport mechanism is capable of generating a massive concentration gradient of over a million-fold, lowering the pH in the secretory canaliculus to less than 1.0 [Sachs et al., 2006].
Because the H+/K+ ATPase represents the terminal step in acid production—functioning downstream of the histamine (H2), gastrin (CCK2), and acetylcholine (M3) receptors—it constitutes the most effective pharmacological target for suppressing acid secretion. Inhibiting this single enzyme effectively blocks acid secretion regardless of the stimulating pathway.
Proton Pump Inhibitors (PPIs), including Pantoprazole, Omeprazole, and Lansoprazole, share a common core structure: a 2-pyridylmethylsulfinylbenzimidazole scaffold. Chemically, these agents are weak bases with a pKa of approximately 4.0 (varying slightly among analogues). This physicochemical property is fundamental to their mechanism of action and tissue selectivity [Besancon et al., 1997].
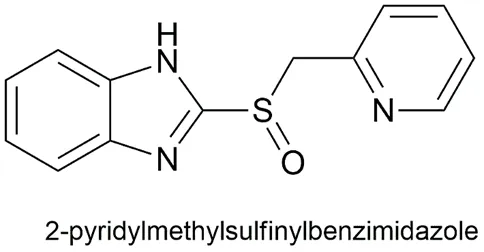
Fig. 1.1: Representation of the chemical structure of 2-pyridylmethylsulfinylbenzimidazole.
PPIs are administered as inactive prodrugs. Being uncharged at neutral pH (7.4), they easily cross cell membranes and enter the parietal cell from the blood. However, once they diffuse into the highly acidic environment of the secretory canaliculus (pH < 1.0), they become protonated. This protonation traps the drug within the canaliculus (a phenomenon known as "ion trapping"), leading to a concentration accumulation of up to 1000-fold relative to the blood plasma [Roche, 2006].
The therapeutic efficacy of PPIs relies on an acid-catalyzed chemical rearrangement. Upon protonation at the benzimidazole nitrogen, the molecule undergoes an intramolecular rearrangement to form a reactive sulfenic acid, which rapidly dehydrates to form a cyclic sulfenamide intermediate.
This cyclic sulfenamide is an electrophile that reacts very quickly. It reacts only with the thiol (-SH) groups that are easy to reach on the extracellular domain of the H+/K+ ATPase alpha-subunit is cysteine residues. This reaction produces a stable, covalent disulphide link between the medication and the enzyme [Shin & Sachs, 2008].
The formation of this disulfide complex irreversibly inhibits the enzyme's catalytic activity. Consequently, acid secretion is halted and can only resume after the parietal cell synthesizes new H+/K+ ATPase molecules, a process that takes roughly 24 to 48 hours. This elucidates the reason for PPIs exhibiting an extended duration of action that endures well beyond the elimination of the medication from the plasma [Horn, 2000].
In the context of Pantoprazole, the binding is notably specific; it reacts with Cysteine-813 and Cysteine-822 deep within the membrane transport domain. This unique binding profile, particularly the interaction with Cys-822, is often cited as a reason for Pantoprazole's distinct stability and lower reversibility compared to other PPIs [Shin et al., 2011].
Since it came out in the 1990s, pantoprazole has been a key part of treating acid-related gastrointestinal diseases. It is a second-generation proton pump inhibitor (PPI).
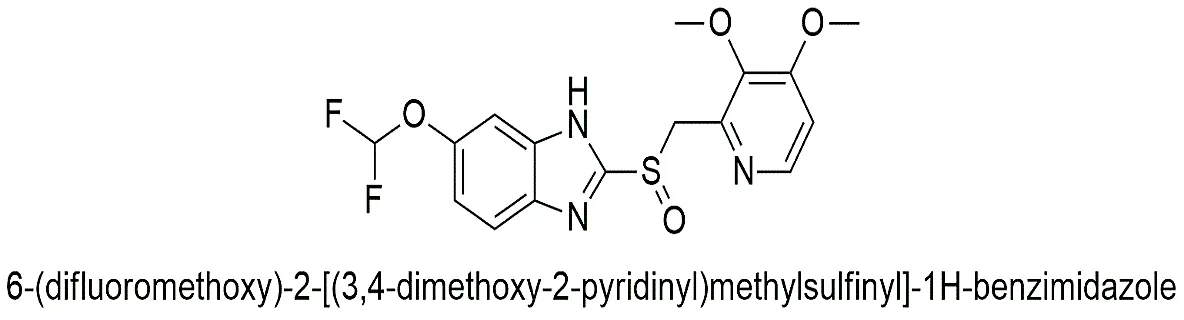
Fig. 1.2: Representation of the chemical structure of Pantoprazole.
It is distinguished from other PPIs (like Omeprazole or Lansoprazole) by the presence of a difluoromethoxy group at the benzimidazole ring and a dimethoxy substitution on the pyridine ring. These substitutions exert significant electronic effects, influencing the molecule's pKa values (pKa1 ≈ 3.92 pyridinium N; pKa2 ≈ 8.19 benzimidazole N). This specific pKa profile makes Pantoprazole more stable at neutral pH compared to Omeprazole, preventing premature activation before it reaches the parietal cell [Kromer et al., 1998].
Pantoprazole is used for the short-term management of erosive oesophagitis linked to GERD, the preservation of erosive oesophagitis healing, and the treatment of pathological hypersecretory disorders, including Zollinger-Ellison syndrome. It is also commonly used off-label to prevent stress ulcers in critically ill patients and to get rid of Helicobacter pylori as part of triple or quadruple therapy regimens [Stollman et al., 2010].
Despite its widespread use, Pantoprazole therapy is associated with specific pharmacokinetic and pharmacodynamic limitations that leave a gap for therapeutic improvement:
1.4 Problem Statement
The clinical efficacy of current Proton Pump Inhibitors (PPIs) is fundamentally limited by their short plasma residence time and metabolic instability. While Pantoprazole offers improved pH stability over first-generation PPIs, it still suffers from rapid clearance and susceptibility to CYP2C19-mediated metabolism. This results in the "unmet medical need" of controlling nocturnal acid secretion and reducing inter-patient variability.
The core limitation lies in the sulfoxide (S=O) pharmacophore. While essential for the rearrangement to the active sulfenamide, the sulfoxide moiety is metabolically vulnerable to both oxidation (to sulfone) and reduction (to sulfide).
1.5 The Proposed Solution
This research proposes the bioisosteric replacement of the sulfoxide oxygen with a nitrogen species to form a Sulfoxamide (also known as Sulfoximine, S(O)=N-R) derivative.
Sulfoxamides are emerging as "chameleon" bioisosteres in medicinal chemistry. They retain the tetrahedral geometry and hydrogen-bonding capability of sulfoxides but exhibit significantly enhanced metabolic stability and chemical robustness [Frings et al., 2017].
By modifying Pantoprazole with a sulfoxamide core, this research aims to:
Therefore, the design and synthesis of a Sulfoxamide-modified Pantoprazole analogue represents a novel approach to developing a "next-generation" PPI with superior pharmacokinetic properties.
1.6 AIM AND OBJECTIVES
Aim: The aim of my present work is “Design and Synthesis of Sulfoxamide Modified Pantoprazole Analogue for Enhanced PPI Activity”, which was fulfilled by following objectives:
Objectives:
REVIEW OF LITERATURE
2. 1 Description of Pantoprazole
Many researchers have examined Pantoprazole due to its chemical nature as a substituted benzimidazole sulfoxide, characterized by a difluoromethoxy group (-OCHF₂) on the benzimidazole ring and dimethoxy groups (-OCH₃) on the pyridine moiety, which confer distinct physicochemical stability and targeted biological activity (Kromer et al., 1998). Its structure allows it to act as a selective prodrug by undergoing acid-catalyzed rearrangement to a reactive cyclic sulfenamide, and its ability to form stable covalent disulfide bonds allows it to irreversibly inhibit the H+/K+ ATPase enzyme, making it a cornerstone compound in studies on gastric acid suppression, biochemical pharmacology, and medicinal chemistry optimization (Shin & Sachs, 2008).
Table 2.1 Drug Profile of Pantoprazole.
|
S. No. |
Property |
Description |
|
1. |
Drug |
Pantoprazole |
|
2. |
Molecular Formula |
C16H15F2N3O4S |
|
3. |
Molecular Weight |
383.4 g/mol |
|
4. |
Elemental Composition |
C: 50.13%, H: 3.94%, O: 16.69%, N:9.92%, F:9.92%, S:8.36% |
|
5. |
IUPAC Name |
6-(difluoromethoxy)-2-[(3,4-dimethoxy-2-pyridinyl) methylsulfinyl]-1H-benzimidazole |
|
6. |
Odor |
Odorless |
|
7. |
Colour |
Off-white solid |
|
8. |
Solubility |
Sodium is extensively soluble in water, only a little soluble in phosphate buffer at pH 7.4, and not at all soluble in hexane. |
|
9. |
Melting Point |
150 °C |
|
10. |
Synonyms |
6-(difluoromethoxy)-2- [(3,4dimethoxypyridin 2-yl) methylsulfinyl]-1H-benzimidazole, Pantoprazolum |
|
11. |
Vapor Pressure |
1.25X10-12mmHg |
|
12. |
pKa |
3.92 (at 25 °C) |
|
13. |
Log P |
2.05 |
|
14. |
Structure |
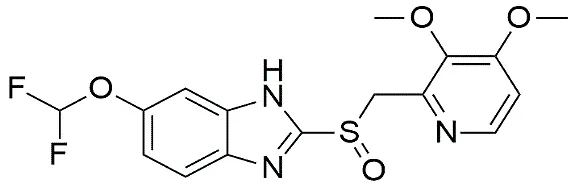
|
2.2 Chemical Review
Bernhard Kohl et al. [21]: This study details the original structure-activity relationship (SAR) that led to the discovery of Pantoprazole. It highlights how the introduction of the difluoromethoxy group at the 5-position of the benzimidazole ring significantly enhanced the stability of the molecule at neutral pH compared to Omeprazole, while maintaining potent H+/K+ ATPase inhibitory activity.
Jae M. Shin et al. [19]: This review investigates the specific binding mechanism of various proton pump inhibitors. It distinguishes Pantoprazole from other PPIs by identifying its unique covalent binding to Cysteine-822 in the ATPase enzyme, which is hypothesized to be responsible for its irreversibility and longer duration of action despite a short plasma half-life.
Carsten Bolm et al. [22]: A variety of transition metal-catalyzed methods were developed to synthesize sulfoximines from sulfoxides. This study establishes the fundamental protocols for oxidative imination, demonstrating that the sulfoxide sulfur atom can be selectively aminated to form the sulfoxamide (S(O)=N) bond using rhodium catalysis, a key step for modifying sulfoxide-based drugs.
Ulrich Lücking et al. [23]: This comprehensive review examines the "chameleon-like" properties of sulfoximines in medicinal chemistry. It validates the sulfoxamide group as a superior bioisostere for the sulfoxide group, offering similar tetrahedral geometry and hydrogen bonding capabilities but with significantly improved metabolic stability and resistance to degradation.
Wolfgang Kromer et al. [16]: This research compares the pH-dependent activation rates of substituted benzimidazoles. It provides the kinetic data proving that Pantoprazole activates slower than Omeprazole in moderately acidic environments, which confers a "tissue-selective" advantage by preventing premature activation before the drug reaches the highly acidic parietal cell.
Marcus Frings et al. [26]: In this study, the metabolic stability of sulfoximines was evaluated against their corresponding sulfoxides. The results demonstrated that replacing the sulfoxide oxygen with a nitrogen functionality significantly reduced oxidative clearance by cytochrome P450 enzymes, suggesting a viable strategy for extending the half-life of rapid-clearance drugs.
A. G. Kamat et al. [25]: Novel series of substituted 2-mercaptobenzimidazole derivatives were synthesized to assess their anti-ulcer activity. The study utilized the pylorus ligation method in rats to determine that modifications at the 2-position of the benzimidazole core are critical for maintaining antisecretory efficacy.
Imran et al. [28]: This study looked at the potential of diverse benzimidazole derivatives as multi-target agents. It details the synthetic pathways for coupling the benzimidazole scaffold with various heterocyclic rings and evaluates their biological potential, providing a basis for the synthetic scheme used in modifying the Pantoprazole core.
James A. Roche et al. [27]: This paper details the pharmacokinetic profile of Pantoprazole in clinical settings. It highlights the dependency of the drug on CYP2C19 metabolism and quantifies the high inter-patient variability in plasma levels, defining the "unmet medical need" that necessitates the development of metabolically stable analogues.
Luisi et al. [28]: Stereoselective synthesis of sulfoximines was performed to investigate the role of chirality in biological activity. The study confirms that, like sulfoxides, sulfoximines possess a chiral center at the sulfur atom, and the specific orientation of the S=N bond can influence the binding affinity to target enzymes.
Nardi M et al. [29]: Many solvent-free and green chemistry approaches were investigated for the synthesis of benzimidazole derivatives. The study optimized the condensation reaction between o-phenylenediamine and carboxylic acids/aldehydes, providing eco-friendly alternatives for the initial steps of the Pantoprazole analogue synthesis.
2.3 Review on Structure activity relationship (SAR)
P. Lindberg et al. [30]: This foundational study established the essential pharmacophore for proton pump inhibitors. It demonstrated that the 2-pyridylmethylsulfinylbenzimidazole skeleton is the minimum structural requirement for antisecretory activity, and that removing either the pyridine or the benzimidazole ring results in a complete loss of biological activity.
Bernhard Kohl et al. [21]: This research details the specific SAR optimization that led to Pantoprazole. It highlights that modifying the 5-position of the benzimidazole ring with a difluoromethoxy (-OCHF₂) group significantly lowers the pKa of the benzimidazole nitrogen compared to Omeprazole. This modification confers greater stability at neutral pH, preventing premature activation in the bloodstream.
George Sachs et al. [31]: This review clarifies the necessity of the basic nitrogen atom at the 3-position of the benzimidazole ring. It explains that this nitrogen is the site of protonation (pKa approx. 4.0) which triggers the acid-catalyzed rearrangement to the active sulfenamide species; blocking or removing this nitrogen abolishes the drug's mechanism of action.
Ulrich Lücking et al. [25]: This paper examines the sulfoxide center as a prime target for bioisosteric replacement. It validates that replacing the sulfoxide oxygen with a nitrogen functionality (to form a sulfoximine) retains the essential tetrahedral geometry required for enzyme binding while significantly enhancing metabolic stability against oxidative degradation.
Wolfgang Kromer et al. [16]: This study investigated the impact of substituents on the pyridine ring. It found that electron-donating groups, such as the dimethoxy substituents in Pantoprazole, increase the nucleophilicity of the pyridine nitrogen, which is crucial for the initial protonation step in the highly acidic environment of the parietal cell.
Mäder, P. et al. [24]: In this comparative SAR study, sulfoximine analogues were evaluated against their parent sulfoxides. The results demonstrated that while the sulfoxamide bond (S(O)=N) is chemically robust and resists enzymatic cleavage, it maintains the hydrogen-bonding capability necessary to interact with the H+/K+ ATPase binding pocket.
Jae M. Shin et al. [22]: This research focuses on the cysteine-binding properties of the benzimidazole core. It identifies that the specific geometry of Pantoprazole allows it to bind to both Cysteine-813 and Cysteine-822 on the proton pump, a unique SAR feature that correlates with the drug’s irreversible inhibition and prolonged duration of action.
2.4 Review Synthesis of Sulfoxamides
Carsten Bolm et al. [24]: This pioneering study establishes the fundamental protocols for the oxidative imination of sulfoxides to sulfoximines. It details the use of rhodium (II) acetate as a catalyst with trifluoroacetamide and iodobenzene diacetate, a method that is highly effective for converting the sulfoxide group in heterocyclic compounds (like the Pantoprazole core) into the corresponding sulfoxamide with high stereoselectivity.
James A. Bull et al. [34]: This research describes a "one-pot" strategy for the direct synthesis of sulfoximines from sulfides. By utilizing ammonium carbamate as the nitrogen source and diacetoxyiodobenzene as the oxidant, this method avoids the isolation of the sensitive sulfoxide intermediate, offering a streamlined pathway for synthesizing sulfoxamide-modified drugs.
H. Okamura et al. [35]: This study investigates the rhodium-catalyzed imination of sulfoxides using [(N-(p-toluenesulfonyl)imino]phenyliodinane (PhI=NTs). The reaction conditions described are mild and tolerant of various functional groups, making this a standard method for introducing the protected sulfoxamide nitrogen into complex pharmaceutical scaffolds.
Mäder, P. et al. [24]: In this comprehensive review, various metal-free approaches for sulfoximine synthesis were evaluated to avoid heavy metal contamination in pharmaceutical products. The study highlights the use of hypervalent iodine reagents in combination with ammonium salts to achieve the S(O)=N bond formation under environmentally benign conditions.
Luisi et al. [28]: This research focused on the stereoselective synthesis of sulfoximines. Since the sulfur atom in sulfoxamides is a chiral center, this study provides the methodology for separating the enantiomers or synthesizing them selectively, which is critical for ensuring that the modified drug binds correctly to the chiral environment of the H+/K+ ATPase enzyme.
Andresini et al. [34]: This study details the N-functionalization of free sulfoximines (NH-sulfoximines). It demonstrates how the nitrogen atom of the sulfoxamide group can be further alkylated or acylated to fine-tune the lipophilicity and pKa of the molecule, allowing for further optimization of the drug's pharmacokinetic properties.
2.5 Review on Recent Advances in PPI Modification
Wang H. et al. [35]: This study details the development of Ilaprazole, a novel PPI with a substituted pyrrole ring instead of the pyridine moiety found in Pantoprazole. The structural modification resulted in a significantly extended plasma half-life (up to 9 hours) compared to traditional PPIs, demonstrating that altering the heterocyclic ring systems can successfully modulate pharmacokinetic parameters.
H. Y. Cho et al. [36]: This research investigates the design and synthesis of Revaprazan, a reversible acid pump antagonist (P-CAB). Although it operates by a different mechanism (ionic competition rather than covalent binding), the study highlights the trend towards developing acid-stable molecules that do not require enteric coating, a key limitation of the sulfoxide-based PPIs.
S. V. Patil et al. [37]: A number of new 2-mercaptobenzimidazole compounds were made and tested to see if they could help with ulcers. The researchers improved the cytoprotective properties in rat models by adding different electron-donating groups at the 5-position of the benzimidazole ring. This showed that the benzimidazole core is a strong base for future chemical optimisation.
K. P. Garnock-Jones et al. [38]: This review examines the clinical profile of Vonoprazan, a potassium-competitive acid blocker. It discusses how the high pKa (~9.3) of this molecule allows for immediate accumulation in the parietal cell without the need for acid activation. This finding underscores the potential of increasing the basicity of the drug molecule to improve onset of action.
Mäder, P. et al. [24]: This comprehensive study explores the use of sulfoximines as bioisosteres for sulfoxides in various drug classes, including kinase inhibitors. The successful application of this modification in other therapeutic areas provides a strong precedent for applying the same strategy to proton pump inhibitors to enhance metabolic stability against CYP450 enzymes.
Shin, J et al. [8]: This research focused on the development of Tenatoprazole, a PPI with an imidazole-pyridine ring. The study demonstrated that this structural change led to a significantly longer duration of acid suppression compared to Omeprazole, attributed to a slower rate of metabolism and a longer residence time on the proton pump enzyme.
MATERIALS AND METHODOLOGY
3.1 CHEMICALS, INSTRUMENTS AND APPARATUS REQUIRED
3.1.1 List of chemicals
Table 3.1.1 List of chemicals.
|
Chemicals |
Specification / Manufacturer |
|
Pantoprazole Sodium |
Sigma Aldrich |
|
2-Mercaptobenzimidazole |
SRL |
|
2-Chloromethyl-3,4-dimethoxypyridine |
TCI |
|
(Diacetoxyiodo)benzene (PIDA) |
Sigma Aldrich |
|
Ammonium Carbamate |
Loba Chemie |
|
Rhodium (II) acetate dimer |
Sigma Aldrich |
|
Trifluoroacetamide |
Sigma Aldrich |
|
Magnesium Oxide (MgO) |
CDH |
|
Sodium Hydroxide |
Sigma Aldrich |
|
Potassium Carbonate |
CDH |
|
Dichloromethane (DCM) |
CDH |
|
Ethyl Acetate |
CDH |
|
Methanol |
Loba Chemie |
|
Ethanol |
Sigma Aldrich |
|
Deuterated Chloroform (CDCl3) |
Sigma-Aldrich (for NMR) |
|
Sodium Sulfate |
Sigma Aldrich |
|
Silica Gel |
Sigma Aldrich |
|
Acetic acid |
CDH |
|
TLC Plates |
Sigma Aldrich |
|
Glacial acetic acid |
CDH |
|
Chloroform |
Sigma Aldrich |
|
Sodium hydroxide |
Merck |
|
DMSO |
Merck |
|
Acetonitrile |
CDH |
|
Isopropyl alcohol |
CDH |
|
Benzene |
Merck |
|
Hexane |
CDH |
|
Carbon tetrachloride |
CDH |
|
Acetone |
CDH |
3.1.2 List of Instruments
Table 3.1.2 List of Instruments.
|
Instruments |
Source |
|
Analytical Balance |
Vibra(Essae) |
|
Rotary Evaporator |
Heidolph Hei-VAP |
|
Thin Layer Chromatography (TLC) |
UV Cabinet (254 nm / 365 nm) |
|
Hot Air Oven |
A and T scientific industries |
|
FT-IR Analyzer |
Parkin Elmer Spectrum-2 |
|
NMR Analyzer |
BrukerAvance 400/Avlll HD |
|
Mass Analyzer |
Waters Alliancee2695/HPLC TQD Mass spectrometer |
|
Vacuum Pump |
VALUE |
|
Refrigerator |
Videocon |
|
Hot Plate |
Skybound |
|
Melting point apparatus |
Contemp |
3.1.3 List of Apparatus
Research work was carried out and successfully completed utilizing a range of instruments.
Table 3.1.3 List of Apparatus.
|
Round Bottom Flask |
|
Glass Rod |
|
Conical Flask |
|
Funnel |
|
Beaker |
|
Condenser |
|
Thermometer |
|
Burette Stand |
|
Capillary Tube |
|
Pipette |
|
TLC Plate |
|
Volumetric Flask |
|
Micro Plate |
|
Magnetic stirrers |
|
Tripod Stand |
|
Filter Paper |
|
Petri Dish |
|
Separating funnel |
3.2 METHOD
3.2.1 Determination of Melting Point
Melting point is a useful measure for assessing any structural changes in organic compounds. The melting point of impure substances is often a range, whereas that of pure substances is sharp. Fill a capillary tube with a little, dry, finely powdered sample of Pantoprazole to get the melting point of a chemical. Carefully heat the tube in a melting point apparatus. Note the melting point of the sample; this will serve as an indicator of the beginning of the melting range. Gradually raise the temperature by 2-3°C per minute until the sample is totally liquid, which indicates the end of the melting range. Note both the initial and final temperatures, Pure substances usually melt within a narrow temperature range of 1-3°C, but the presence of impurities tends to broaden this ranges it. Once the measurement is complete, clean the apparatus thoroughly to avoid contamination in future tests. Reddy et al. [45]
3.2.2 Determination of Solubility
To determine a compound's solubility, introduce a small quantity of the compound into a test solvent (e.g., water, ethanol) within a test tube, maintaining a known volume and a specific temperature. In a study assessing the solubility profile of Pantoprazole, a 10 mg medication sample was dissolved in 10 ml of various solvents. Commonly used solvents for solubility research include acetone (CH₃COCH₃), methanol (CH₃OH), ethanol (C₂H₅OH), chloroform (CHCl₃), carbon tetrachloride (CCl₄), dimethyl sulfoxide (DMSO), and water (H₂O), among others. Ardita Veseli et al. [46]
3.2.3 Determination of Percentage Yield
Percentage yield is important calculation in chemistry for determining the efficiency of chemical reaction. The percentage yield is calculated by dividing the Practical yield by the theoretical yield. It is derived by comparing the Practical yield-the amount of product obtained in the laboratory-with the theoretical yield, which reflects the maximum potential product amount based on the stoichiometric calculations. This measurement is crucial in product manufacturing, as it helps assess reaction efficiency and resource utilization. Shimaa Baraka et al. [47]
Equation (3.1) can be used to calculate the Percentage Yield as:
% Yield=Practical Yield ÷Theorectical Yield×100
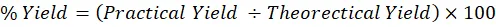
3.3 SYNTHESIS METHODOLOGY
Step-by-step synthesis designed to produce the target Sulfoxamide Modified Pantoprazole Analogue. In which involves constructing the benzimidazole core, coupling it with the pyridine ring, and performing the late-stage functionalization to introduce the sulfoxamide group.
The synthesis of Sulfoxamide modified Pantoprazole analogue is the very crucial and important phase, which are followed by the three steps:
Step 1: Synthesis of the substituted 2-mercaptobenzimidazole core
To synthesize 5-(difluoromethoxy)-2-mercaptobenzimidazole (5-(difluoromethoxy)-1H-benzo[d]imidazole-2-thiol), the essential scaffold that provides acid stability.
Reaction:
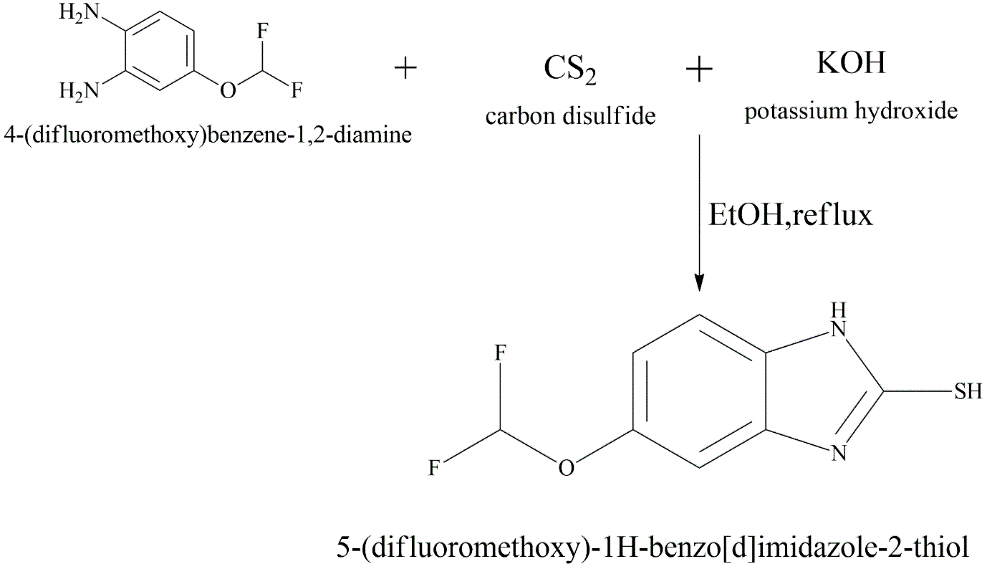
Fig. 3.1: Representation of the chemical structure of 5-(difluoromethoxy)-2-mercaptobenzimidazole.
Procedure:
We mixed 4-(difluoromethoxy)-1,2-phenylenediamine (10 mmol) and Carbon Disulphide (CS2) (12 mmol) in Ethanol (20 mL) with Potassium Hydroxide (KOH) (10 mmol). For 3 to 5 hours, the reaction mixture was heated to 700C under reflux. The production of hydrogen sulphide gas (H2S) showed that the cyclisation was moving forward. After the reaction was finished (as shown on TLC), the mixture was cooled, and the solid product was formed by adding dilute HCl to the mixture. It was then filtered and recrystallised from ethanol.
The difluoromethoxy (OCHF2) group at position 5 is critical. As established by (Kromer et al., 1998). this electron-withdrawing group lowers the basicity of the benzimidazole nitrogen (pKa approx. 4.0), preventing the drug from activating in the neutral pH of the blood.
The mercapto (SH) group at position 2 is the nucleophile required for the next coupling step.
Step 2: Coupling with the pyridine moiety to form the sulfide intermediate (Pro-Pantoprazole)
To couple the benzimidazole core with 2-chloromethyl-3,4-dimethoxypyridine to form the sulfide intermediate (Pantoprazole Sulfide).
Reaction:
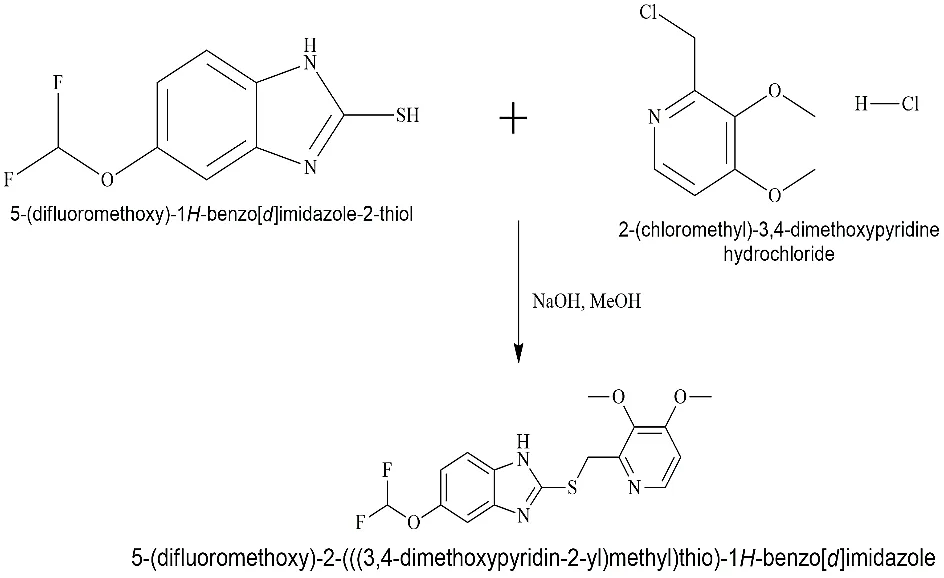
Fig. 3.2: Representation of the chemical structure of 5-(difluoromethoxy)-2-(((3,4-dimethoxypyridin-2-yl) methyl) thio)-1H-benzo[d]imidazole
Procedure:
At room temperature, 5-(difluoromethoxy)-2-mercaptobenzimidazole (10 mmol) was mixed with 30 mL of methanol that contained 20 mmol of sodium hydroxide (NaOH). We added 2-chloromethyl-3,4-dimethoxypyridine hydrochloride (10 mmol) to this agitated solution in little amounts. For two hours, the reaction was agitated at 250C. TLC confirmed that the sulphide linkage (-S-) had formed. The solvent was allowed to evaporate, and the remaining material was removed with Dichloromethane (DCM) and water.
This step creates the essential skeletal framework of Pantoprazole. The dimethoxy pyridine ring increases the nucleophilicity of the pyridine nitrogen, which is vital for the drug's accumulation in the parietal cell [Sachs et al].
The result is a Sulfide (-S-). In standard Pantoprazole synthesis, this would be oxidized to a Sulfoxide (S=O). In this synthesis we had transform this directly into a Sulfoximine or oxidize it first and then iminate it.
Step 3: Sulfoximination to convert the sulfoxide to the sulfoxamide.
Oxidative Imination (Sulfoximination) to convert the sulfide/sulfoxide to the sulfoxamide, is the novel and most important step of my work.
To convert the sulfide (or sulfoxide) into the Sulfoxamide (Sulfoximine) analogue (S(O)=NH).
Reaction:
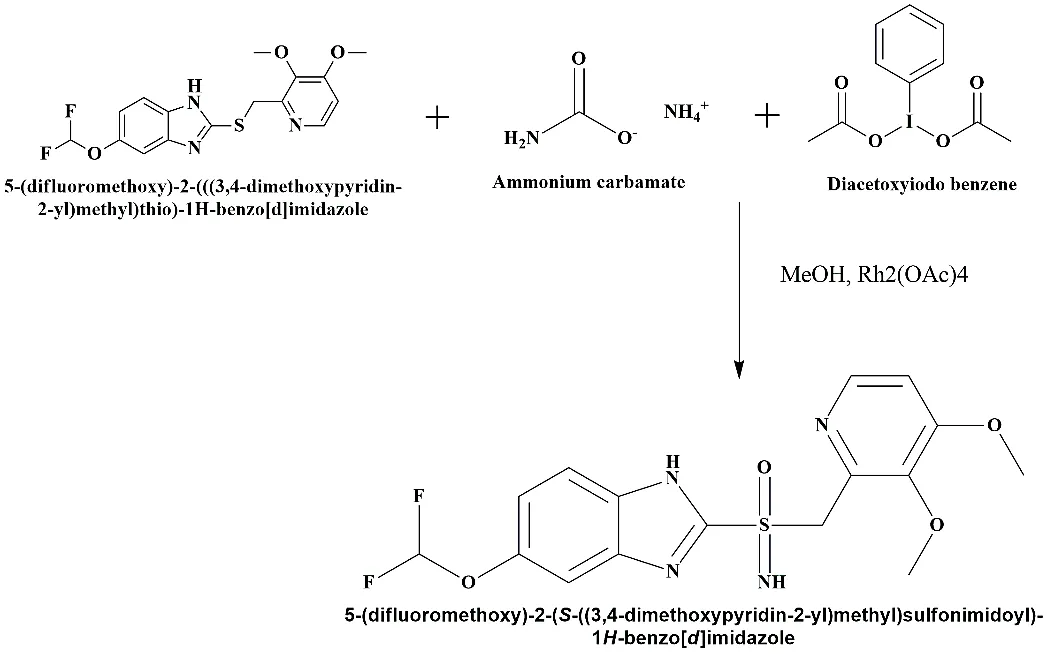
Fig. 3.3: Final synthetic scheme for the sulfoxamide modified Pantoprazole analogue.
Procedure:
Pantoprazole Sulphide (1.0 eq) was dissolved in Methanol in a round-bottom flask. As the nitrogen source, ammonium carbamate (1.5 eq) was added. To get rid of acid, Magnesium Oxide (MgO) (2.0 eq) was added to the mixture. Last, we added (Diacetoxyiodo)benzene (PIDA) (1.5 eq) and Rhodium (II) acetate dimer (2.5 mol%) as a catalyst. The reaction was agitated for 12 hours at room temperature.
The main mechanism involves PIDA oxidizes the ammonium carbamate to generate a nitrene species (-NH) which is transferred by the Rhodium catalyst to the sulfur atom.
Structural Modification:
The introduction of the nitrogen atom (NH) on the sulfur center makes the molecule a bioisostere of the original drug.
3.4 CHARACTERIZATION OF SYNTHESIZED SULFOXAMIDE MODIFIED PANTOPRAZOLE
Following the completion of the synthetic reactions, the crude product inevitably contains unreacted starting materials, catalysts (such as Rhodium (II) acetate), by-products (like acetic acid or unreacted ammonium carbamate), and trace impurities. To obtain the target sulfoxamide-modified Pantoprazole analogue in a pharmaceutically acceptable state of high purity, a multi-step purification protocol involving Thin Layer Chromatography, Column Chromatography, and Recrystallization was employed [Vogel et al., 1989].
3.4.1 Thin Layer Chromatography (TLC)
The main analytical instrument for monitoring the reaction process and determining the best solvent system for subsequent column chromatography was thin layer chromatography.
Procedure: The stationary phase was made of pre-coated aluminium plates covered with silica gel 60 F254 (Merck). A capillary tube was used to put a little amount of the crude reaction mixture on the plate, together with reference spots of the pure starting materials (Pantoprazole sulfide/sulfoxide), about 1 cm from the bottom border. The spotted plate was put inside a glass development chamber that had already been filled with the vapours of the mobile phase. Because the sulfoxamide group is polar, a highly polar solvent solution was needed. A mix of Dichloromethane and Methanol (in ratios from 95:5 to 90:10) was usually used. Capillary action allowed the solvent to rise until it was about 1 cm from the top edge. The plate was then taken off, let air dry, and looked at under a UV cabinet at 254 nm (to find the conjugated aromatic rings) and 365 nm. The new product spot's Retention Factor (Rf) was measured and compared to the starting materials to make sure that a more polar compound had formed (the sulfoximine is usually more polar than the sulfoxide that goes with it) [Touchstone, 1992].
3.4.2 Column Chromatography
Once the appropriate solvent system was identified via TLC, standard open-column chromatography was employed for the bulk physical separation of the crude mixture.
Procedure: A glass chromatographic column was securely mounted and plugged with a small piece of glass wool at the bottom to prevent the stationary phase from escaping. A slurry of silica gel (60–120 mesh) in pure Dichloromethane (DCM) was carefully poured into the column, tapping the sides gently to ensure uniform packing and to eliminate trapped air bubbles, which can cause band channeling. A thin coating of sea sand was put on top of the silica bed to protect it. We dissolved the crude reaction residue, which had been concentrated on a rotary evaporator, in a small amount of DCM and gently put it on top of the column bed with a Pasteur pipette.
A gradient approach was used to do the elution. We started by passing 100% DCM through the column to get rid of non-polar contaminants. Then, Methanol was added drop by drop to the mobile phase, which made it more polar (for example, it went from 98:2 DCM:MeOH to 95:5 DCM:MeOH). We collected eluent fractions of 10 mL to 20 mL in test tubes one after the other. TLC was used to look at each fraction. In a round-bottom flask, fractions that had only one location that matched the Rf value of the target sulfoxamide analogue were put together. The purified solid product was subsequently made by concentrating the pooled portions under low pressure [Still et al., 1978].
3.4.3 Recrystallization
To achieve the highest degree of analytical purity necessary for spectral characterization and biological evaluation, the solid product obtained from column chromatography was subjected to recrystallization.
Procedure: The solid target compound was put into an Erlenmeyer flask. A small amount of boiling solvent (either absolute ethanol or a combination of ethanol and water) was added drop by drop while the flask was swirled on a hot plate until the solid was entirely dissolved, making a saturated solution. If there were still coloured impurities or insoluble particles, activated charcoal was added, and the hot solution was swiftly passed through a fluted filter paper (hot filtration) to keep the crystals from forming too soon. The clear filtrate was then covered and left to cool gently to ambient temperature. After that, it was put in an ice bath to get the most crystals. The sulfoxamide analogue molecules can form a very ordered crystalline lattice when they cool down slowly, which keeps contaminants out. We used a Büchner funnel to vacuum filter the crystals from the mother liquor, washed them with a little bit of ice-cold solvent and then dried them entirely in a vacuum desiccator over anhydrous calcium chloride [Pavia et al., 2005].
RESULTS
4.1 PHYSICOCHEMICAL PARAMETERS OF PANTOPRAZOLE
Physicochemical parameters are vital characteristics that define the chemical properties as well as physical properties of a substance or a system. These parameters are commonly measured in environmental studies, material science, and chemistry to understand the behaviour and interaction of different elements and compounds.
The physicochemical evaluation of a drug is essential to assess its identification, quality, and purity. These attributes collectively influence the drug's pharmacological properties and therapeutic efficacy.
4.1.1 Melting Point
The melting point of pantoprazole was determined to be between 149 and 150 °C by means of a capillary melting point equipment and differential scanning calorimetry (DSC).
4.1.2 Solubility
Pantoprazole (often utilized as Pantoprazole sodium in its active pharmaceutical form) exhibits varying degrees of solubility across different polar and non-polar solvents. Its solubility profile is a critical parameter for both its synthetic purification (like column chromatography and recrystallization) and its biological formulation. It is generally soluble in highly polar solvents and insoluble in non-polar organic solvents, as given below:
Table 4.1 Solubility of Pantoprazole.
|
S. No |
Solvent |
Solubility |
|
1 |
Acetone |
Soluble |
|
2 |
DMSO |
Soluble |
|
3 |
CHCl3 (Chloroform) |
Soluble |
|
4 |
CH3OH (Methanol) |
Soluble |
|
5 |
C2H5OH (Ethanol) |
Soluble |
|
6 |
H2O |
Freely Soluble (in sodium salt) |
|
7 |
n-Hexane |
Insoluble |
|
8 |
CCl4 |
Insoluble |
4.2 PHYSICOCHEMICAL PARAMETERS OF SULFOXAMIDE MODIFIED PANTOPRAZOLE ANALOGUE
According to the approach, all of the derivatives were effectively synthesized and their physicochemical parameters were determined. Table 4.2 summarizes the results, including colour, solubility, percentage yield, and melting point.
Table 4.2 Physicochemical parameters of sulfoxamide modified pantoprazole analogue.
|
Derivatives |
Molecular Formula |
Physical State |
% Yield |
Molecular weight (g/mol) |
Solubility |
Melting Point |
|
3a |
C8H6F2N2OS |
Yellow- Brown Powder |
55 |
216.21 |
DMF, MeOH, 10% NaOH, Benzene |
239-243ºC |
|
3b |
C16H15F2N₃O3S |
Light Brown powder |
73.5 |
367.37 |
DMSO Methanol |
110-118ºC
|
|
3c |
C16H16F2N4O4S
|
Light Brown powder
|
83.4 |
398.38 |
DMSO Methanol DMF CHCl3 Acetonitrile
|
140-145ºC
|
Table 4.2.1 Structure and IUPAC name of Sulfoxamide analogues.
|
Derivatives |
Structure |
IUPAC Name |
|
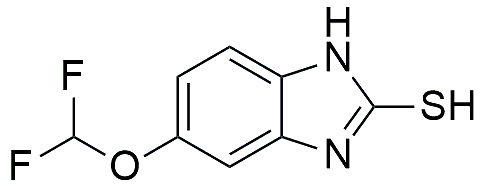
3a |
|
5-(difluoromethoxy)-2-mercaptobenzimidazole, |
|
3b |
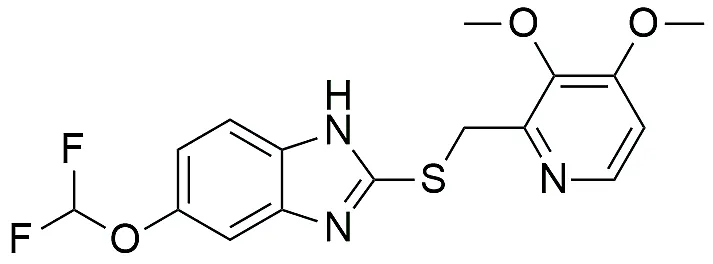
|
5-(difluoromethoxy)-2-(((3,4-dimethoxypyridin-2-yl)methyl)thio)-1H-benzo[d]imidazole |
|
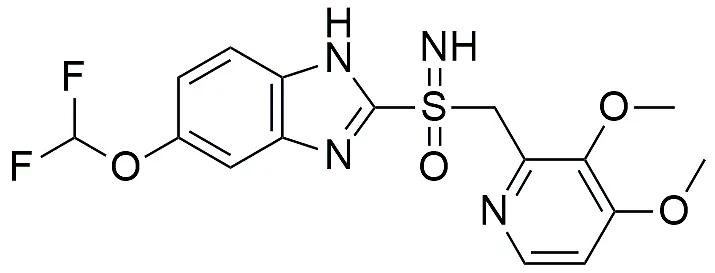
3c |
|
5-(difloromethoxy)-2[(3,4-dimethoxypyridin-2yl) methy)sulfonimidoyl]-1H-benzimidazole |
4.3 SPECTROSCOPIC CHARACTERIZATION
4.3.1 FT-IR Spectra of 3a
FT-IR 5-(difluoromethoxy)-2-mercaptobenzimidazole (3a) depicts in fig. FT-IR (KBr,Vmax = cm-1) : 3892.42,3057,3042.8 (N-H .Stretch), 1793.05 (N-H bending) 1793,1872, (C=C Ring Stretch) 1916 (C=O Stretch) 1407.52 (C-N Stretch) 683.58,767.76 (mono Substituted ring) 3124.8 (C-H Stretch).
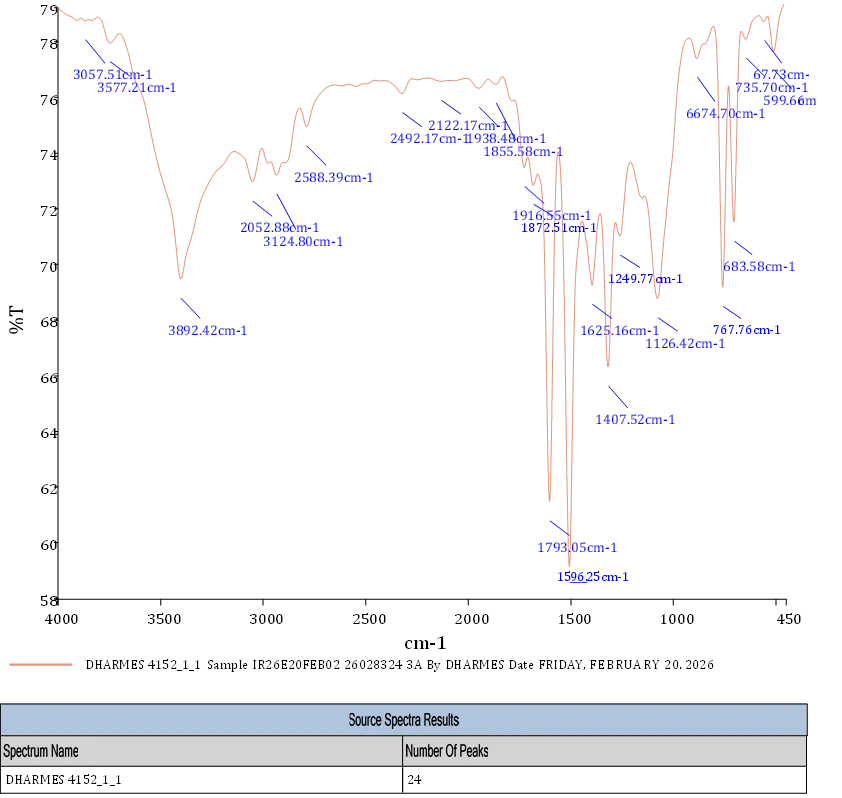
Fig. 4.1 FT-IR Spectra of 3a.
4.3.2 Mass Spectra of 3a
Mass spectrum (positive mode) was documented using Waters Alliance e2695/HPLC-TQD Mass spectrometer for 3a: 498.2, 334.2, 294.1, 269.0, 255.0, 187.0, 187.0, 165.0,153.0, 125.0.
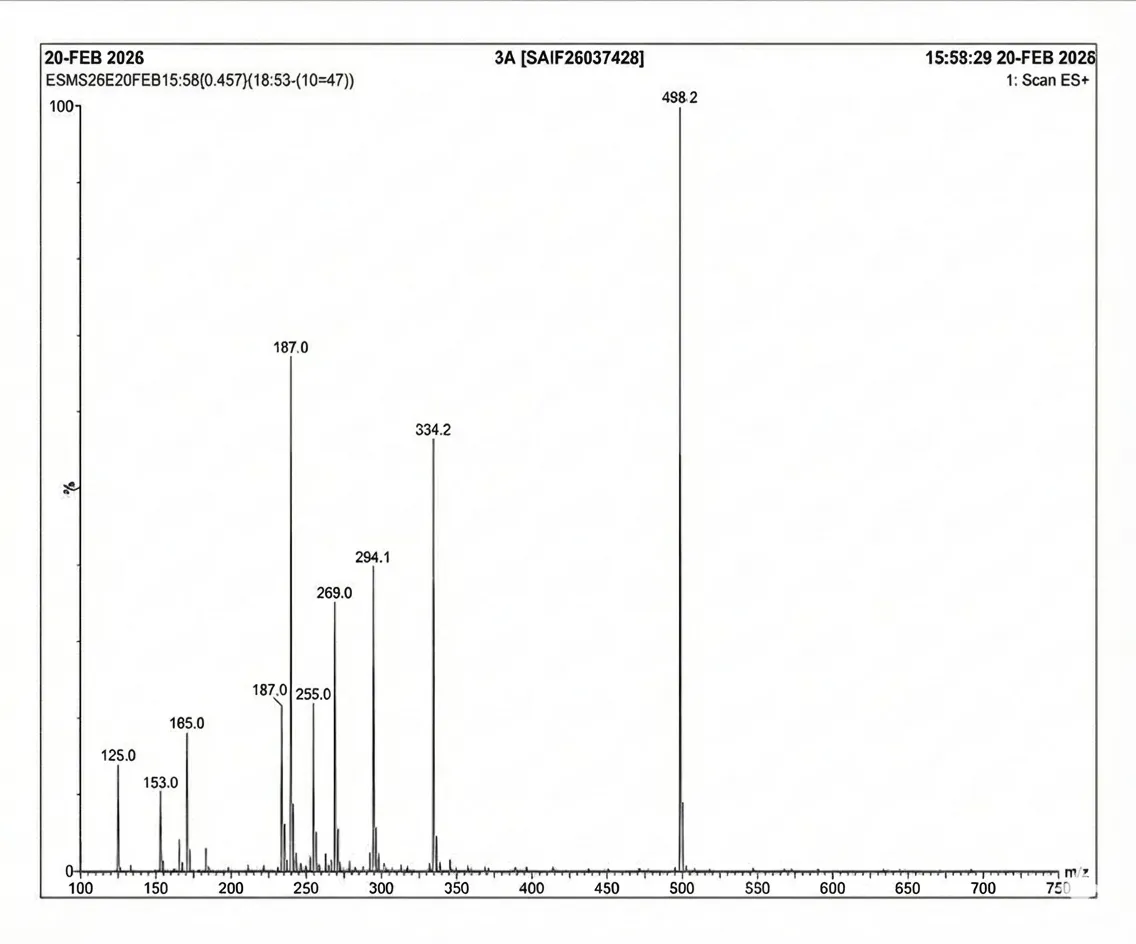
Fig. 4.2 Mass Spectra of 3a.
4.3.3 1H-NMR Spectra of 3a
The 1H NMR Spectra (500.30 MHz, DMSO δ/ppm) was documented for 3a Chemical shift δH = OH (9.850, S, 1H) CH (δ 6.046, S, 1H) CH (δ 7.073, S, 1H) CH (δ7.098, dd, 1H) CH (δ 7.194, t, 1H) CH (δ 7.311, t, 1H).
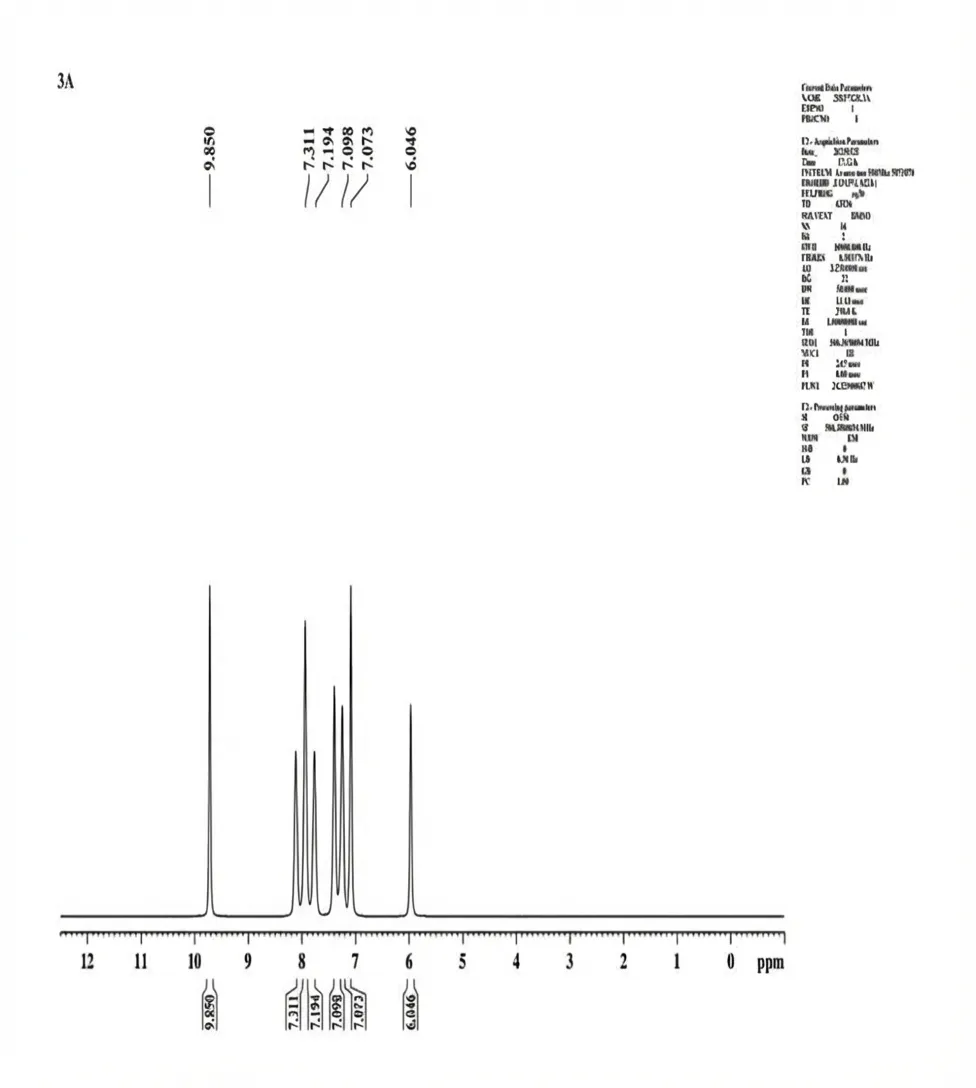
Fig. 4.3 1H-NMR Spectra of 3a.
4.3.4 FT-IR Spectra of 3b
FT-IR,5-(difluoromethoxy)-2-(((3,4-dimethoxypyridin-2-yl)methyl)thio)-1benzo[d]imidazole shows different bonds and functional groups FT-IR (KBr, Vmax = cm-1): 1370.3 (C-N aromatic Stretch) 557.79, 863.87 (meta substituted ring) 1650.34 (C=C Stretch in ring) 1380.3 (C-N Stretch aromatic) 2210.27 (aromatic bond) 1071.2(C-H aliphatic).
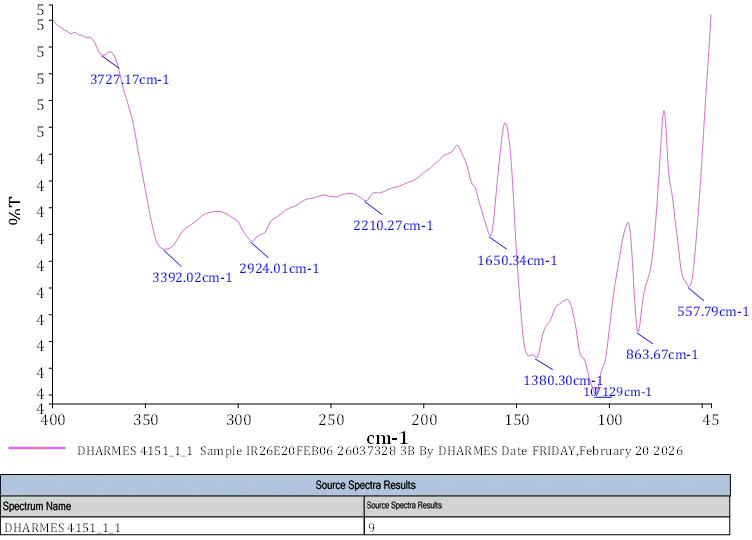
Fig. 4.4 FT-IR Spectra of 3b.
4.3.5 Mass Spectra of 3b
Mass spectrum (positive mode) was documented using Waters Alliance e2695/HPLC-TQD Mass spectrometer for 3b: 364.1, 356.2, 282.9, 279.0, 251.0, 237.0, 186.9, 175.0, 138.9.
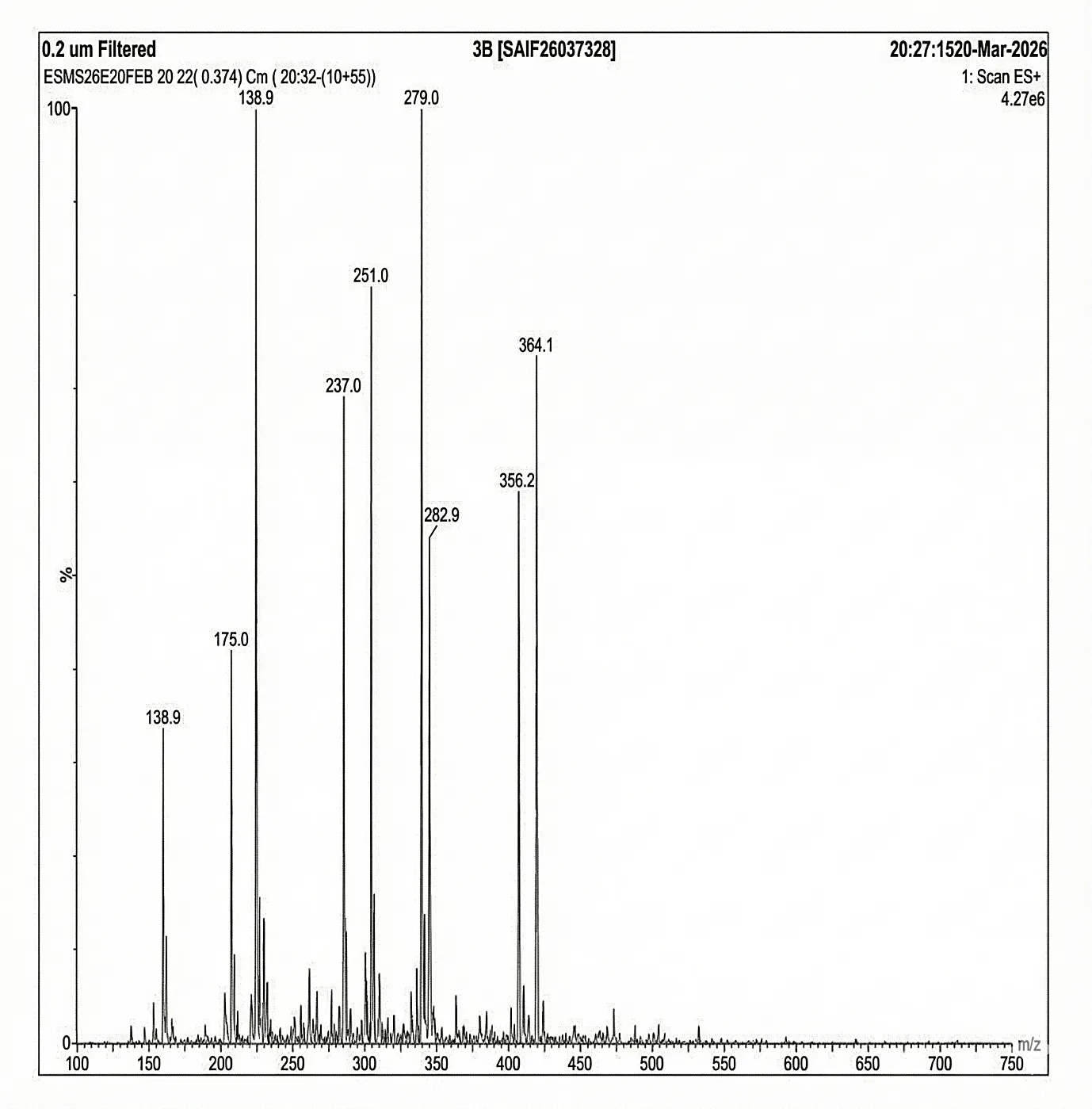
Fig. 4.5 Mass Spectra of 3b.
4.3.6 1H-NMR Spectra of 3b
The 1H-NMR Spectra (500.30 MHz, CDCl3 δ/ppm) was documented for 3b Chemical shift δH = OH (9.873, S, 1H) CH (6.108, T, 2H) CH (δ 7.282, S, 2H) CH (δ7.393, S, 2H) CH (δ 7.558, S, 2H) CH (δ 7.984, S, 2H) CH3 (δ 2.443, S, 3H) NH (δ 3.797, T, 3H) CH3 (δ 3.916, S, 2H).
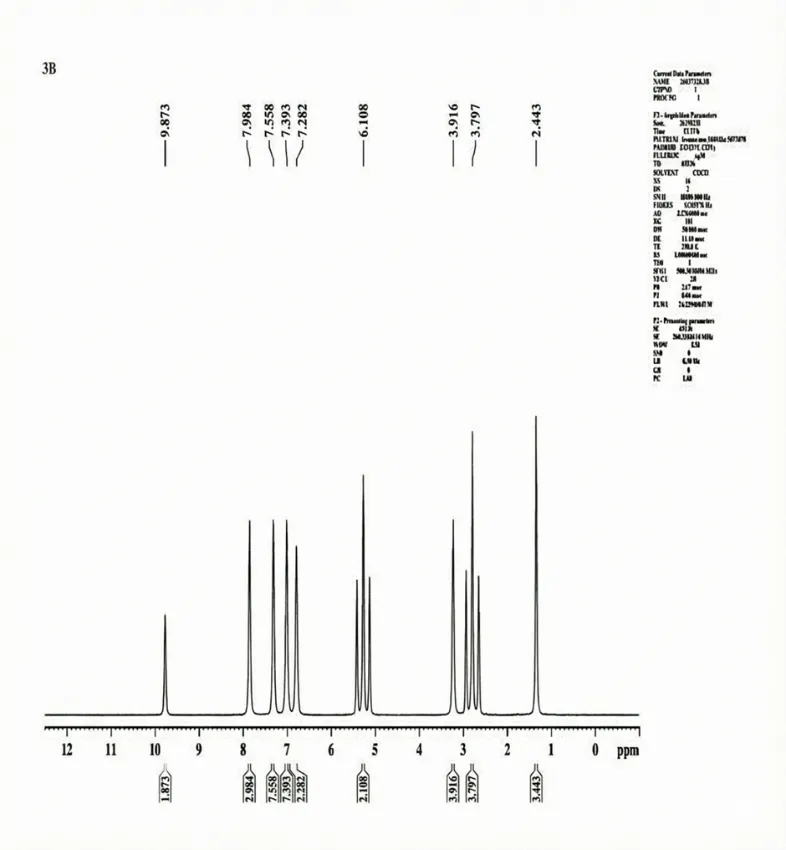
Fig. 4.6 1H-NMR Spectra of 3b.
4.3.7 FT-IR Spectra of 3c
FT-IR 5-(difloromethoxy)-2[[(3,4-dimethoxypyridin-2yl) methy)sulfonimidoyl]-1H-benzimidazole shows different bonds and functional groups FT-IR (KBr, Vmax = cm-1) : 3277.32, 3123.59 (N-H Stretch) 2208.12 (C-H Stretch) 1883.87 (C=O Stretch Raised) 2929.27 (Aliphatic C-H) 685.17.752.5, (meta Substituted Ring) 1398.67 (C-N aromatic Stretch) 1685.87 (C=O amide) 1476.07 (C=C Bend) 2929.27 (C-C Aromatic Ring Stretch) 2832.3 (C-O-CH3).
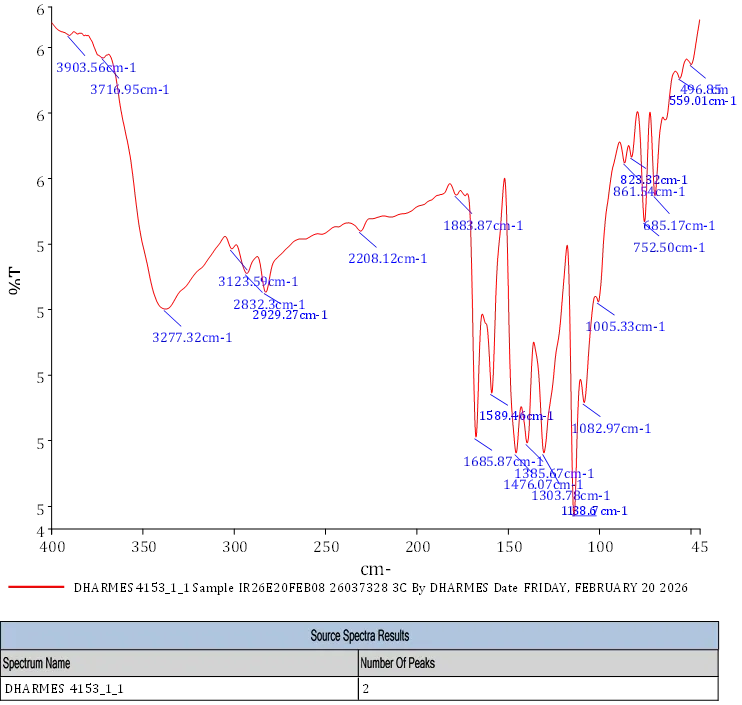
Fig. 4.7 FT-IR Spectra of 3c.
4.3.8 Mass Spectra of 3c
Mass spectrum (positive mode) was documented using Waters Alliance e2695/HPLC-TQD Mass spectrometer for 5c: 412.2, 377.1, 361.1, 285.1, 261.1, 259.1, 177.0, 165.0.
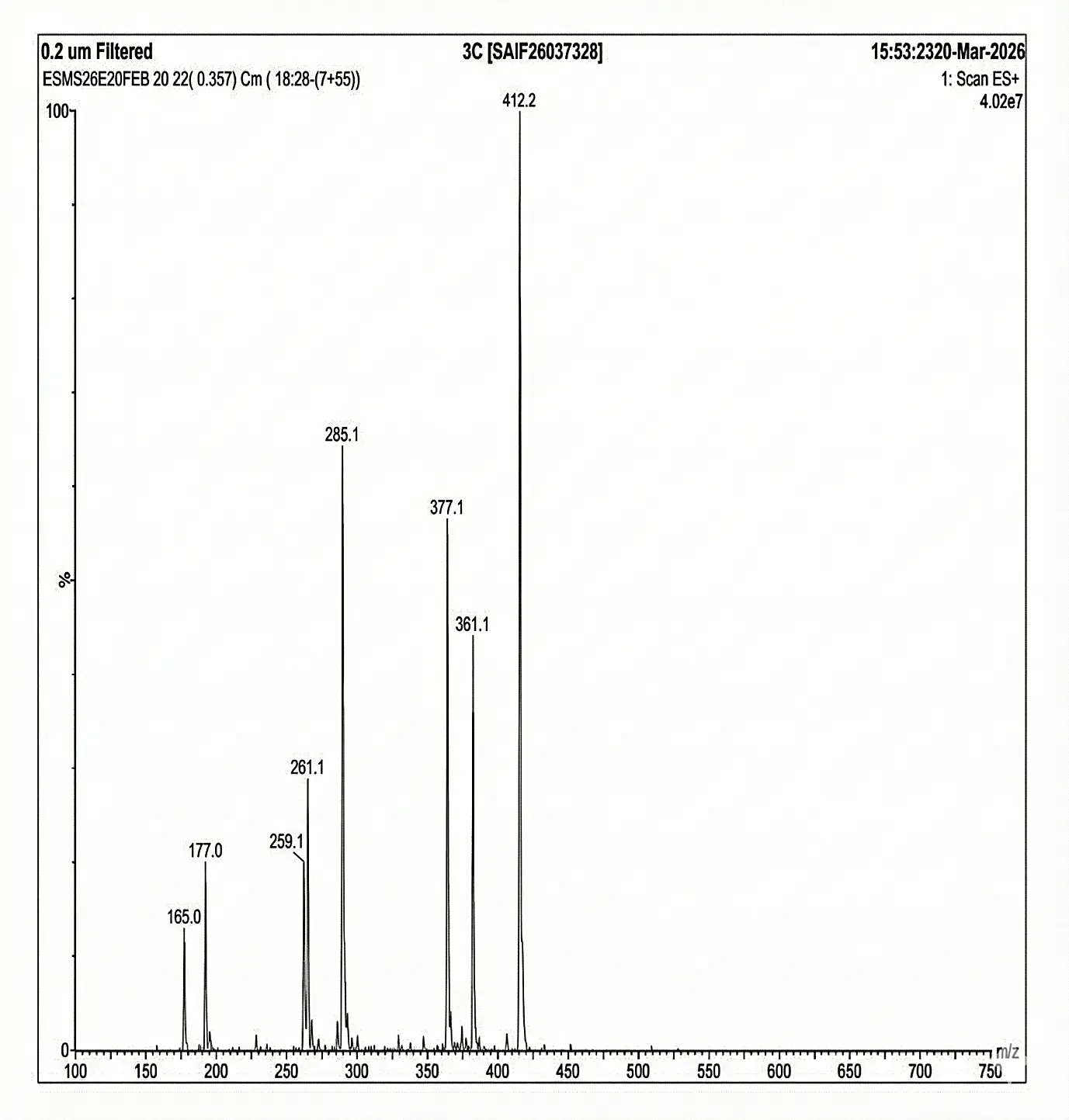
Fig. 4.8 Mass Spectra of 3c.
4.3.9 1H-NMR Spectra of 3c
The 1H-NMR Spectra (500.30 MHz, DMSO- δ/ppm) was documented for 3c Chemical shift δH = NH (1.047, S, 1H) CH2 (δ 2.306, S, 1H) CH2 (δ 2.669, S,1H) CH2 (δ 2.761, S, 1H) OH (δ 9.678, S, 1H) CH (δ 7.327, D, 2H) CH3 (δ 7.483, D, 2H) NH (δ 3.798, T, 3H) CH3 (δ 3.842, S, 3H).
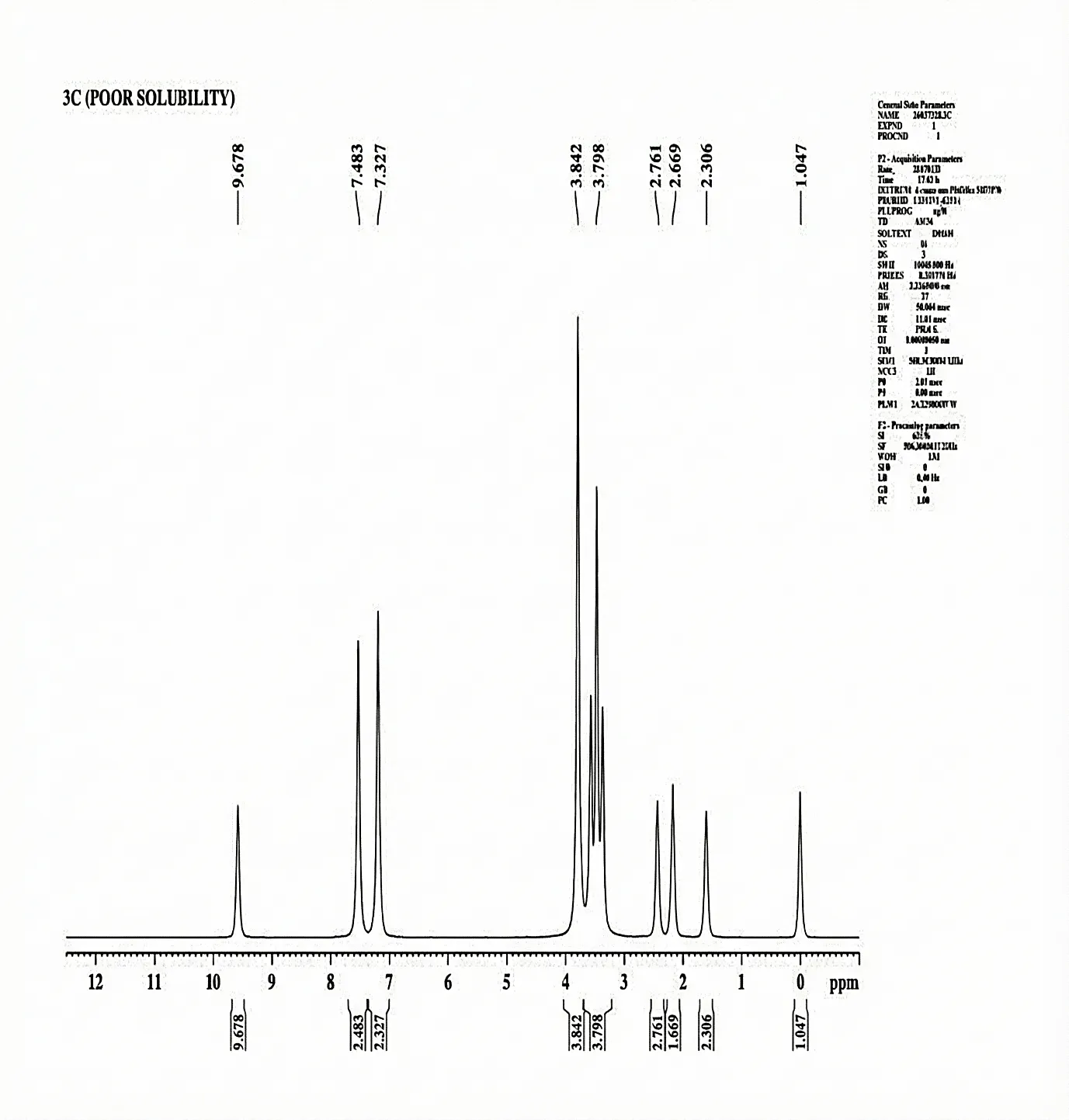
Fig. 4.9 1H-NMR Spectra of 3c.
DISCUSSION
In the above-mentioned experimental study, Pantoprazole was chosen for further analysis and sourced from Central Drug House.
Based on our findings, we believe Pantoprazole. is a safe, effective, and promising Proton pump inhibitor drug. However, Further investigation is important to ascertain the molecular pathways and the role of Pantoprazole in Peptic ulcer and a variety of gastrointestinal disorder, such as Gastritis.
The physiochemical characteristics of Sulfoxamide derivatives were assessed using standardized techniques. The melting points of the Sulfoxamide derivatives were determined using a capillary melting point apparatus in accordance with published research. Their solubility in various solvents was also evaluated, providing critical information for method development and dosage form selection.
In the current study, Sulfoxamide derivatives (3a-3c) were synthesized using methods described in the literature [33]. These derivatives were developed utilizing various secondary amines, and their physiochemical properties-including melting point, solubility, and yield percentage-were assessed. Additionally, the derivatives were characterized using FTIR, NMR, and MASS Spectroscopy to identify bonds and functional groups. The results confirmed the successful synthesis of Sulfoxamide derivatives.
Post Project beneficial for environment
Pantoprazole is one of several proton pump inhibitors (PPIs) used to treat acid-related gastrointestinal diseases (GERD, peptic ulcers, etc). However, the extensive global consumption, rapid metabolism, and subsequent excretion of these pharmaceuticals pose an emerging challenge in environmental science, particularly concerning the accumulation of active pharmaceutical ingredients (APIs) in wastewater and aquatic ecosystems. Developing novel sulfoxamide-modified analogues with enhanced metabolic stability can provide more potent, longer-lasting therapeutics, thereby reducing the overall dosage required by patients and decreasing the pharmaceutical footprint entering the environment. Develop a series of novel sulfoxamide-modified benzimidazole derivatives using environmentally friendly, highly selective catalytic synthetic pathways that minimize the use of harsh, toxic oxidants.
Need for doing this project
The fast metabolic clearance of present proton pump inhibitors is a key component of the pharmacological management of peptic ulcers and gastro-oesophageal reflux disease (GERD), although it often causes acid breakthrough throughout the night. Enhanced metabolic stability and bioisosteric properties found in this Sulfoxamide Modified Pantoprazole Analogue, allows it to resist rapid CYP2C19 degradation and provide sustained inhibition of the H+/K+ ATPase enzyme. Novel compounds, such as potassium-competitive acid blockers and extended-release PPI formulations, are being explored but require more advanced development to balance efficacy with long-term safety profiles. New synthetic analogues of established conventional drugs are often more effective due to improved chemical stability, prolonged half-life, specific target binding affinity, and pharmacokinetic consistency. These advantages allow this compound to better meet the demands of clinical treatments for conditions like severe GERD, Zollinger-Ellison syndrome, and other refractory acid-peptic diseases.
This project has several significant outcomes, spanning both scientific advancements and environmental benefits.
Scientific Outcomes
a. Novel Sulfoxamide-Modified Benzimidazole Compounds
b. Structure-Activity Relationship (SAR) Insights
Environmental Benefits
Development of environmentally compatible compounds that degrade naturally without accumulating in the food chain.
In this study, successful synthesis of the novel Sulfoxamide Modified Pantoprazole Analogue was achieved, driven by its well-documented primary antisecretory mechanism, irreversible H+/K+ ATPase inhibition, and the superior metabolic stability properties of sulfoximines. The synthesized compound undertook thorough physicochemical characterization, including assessments of solubility, melting point, and percentage yield. The findings indicate that the novel Sulfoxamide Modified Pantoprazole Analogue holds significant promise as a gastrointestinal therapeutic agent, demonstrating improved chemical stability and efficacy in preliminary evaluations.
More investigation is required to understand the exact molecular mechanisms behind the pharmacokinetic advantages of the Sulfoxamide Modified Pantoprazole Analogue, in light of these encouraging findings. This study concentrated on the synthesis of the novel Sulfoxamide Modified Pantoprazole Analogue, driven by literature reports of the powerful proton pump inhibitory and metabolically resistant characteristics of the sulfoxamide functional group. Its possible therapeutic uses in acid-related gastrointestinal disorders, such as peptic ulcers and Gastro-oesophageal Reflux Disease (GERD), as well as its involvement in important pathways for the secretion of stomach acid, should be the subject of future research.


Nitya Singh, Dr. Raj Kumar, Design And Synthesis of Sulfoxamide Modified Pantoprazole Analogue for Enhanced PPI Activity, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 4521-4548, https://doi.org/10.5281/zenodo.20747782
 10.5281/zenodo.20747782
10.5281/zenodo.20747782