
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutics, Indore Institute of Pharmacy, Indore, Pithampur Road, Opposite IIM, Indore Madhya Pradesh, 453331
Hypertension is one of the most prevalent cardiovascular disorders worldwide and requires long-term pharmacological therapy for effective blood pressure control. Losartan Potassium, an angiotensin II receptor antagonist, possesses a short biological half-life and relatively low oral bioavailability, making it a suitable candidate for the development of a controlled-release dosage form. The present study was undertaken to formulate and evaluate controlled-release matrix tablets of Losartan Potassium using the direct compression method. HPMC K100M, Eudragit, and Carboxymethyl Cellulose (CMC) were employed as release-retarding polymers to achieve prolonged drug release. The prepared formulations were evaluated for pre-compression parameters, post-compression characteristics, FTIR compatibility studies, drug content, and in vitro dissolution studies. All formulations exhibited satisfactory flow properties, acceptable hardness, friability, and weight variation within pharmacopoeia limits. FTIR analysis confirmed the absence of significant drug–excipient interactions, indicating good compatibility between the drug and selected excipients. Among the prepared formulations, formulation F2 demonstrated the most desirable controlled-release behavior by releasing 98.2 ± 0.5% of the drug over 12 hours while maintaining satisfactory physicochemical properties. The optimized formulation exhibited desirable controlled-release characteristics with satisfactory physicochemical properties, indicating its potential to improve therapeutic efficacy, reduce dosing frequency, and enhance patient compliance in the long-term management of hypertension.
Hypertension is one of the most prevalent chronic cardiovascular disorders and remains a major public health concern worldwide. Persistent elevation of arterial blood pressure significantly increases the risk of cardiovascular diseases, including stroke, myocardial infarction, heart failure, and chronic kidney disease. According to the World Health Organization (WHO), hypertension is one of the leading causes of premature mortality, emphasizing the need for effective long-term pharmacological management.¹
Losartan Potassium is a selective angiotensin II type-1 (AT₁) receptor antagonist widely prescribed for the treatment of hypertension. The drug effectively lowers blood pressure by blocking the vasoconstrictor action of angiotensin II and reducing peripheral vascular resistance. However, Losartan Potassium exhibits relatively low oral bioavailability (approximately 32–35%) due to extensive first-pass metabolism and possesses a short elimination half-life of about 1.5–2.5 hours. Consequently, conventional immediate-release tablets require repeated administration, which may result in fluctuations in plasma drug concentration, reduced therapeutic efficacy, and poor patient compliance.²˒³
Controlled-release drug delivery systems have been extensively investigated to overcome the limitations associated with conventional dosage forms. These systems are designed to release the drug at a predetermined rate over an extended period, thereby maintaining therapeutic plasma concentrations, minimizing peak-to-trough fluctuations, reducing dosing frequency, and improving patient adherence. Hydrophilic matrix tablets prepared using polymers such as Hydroxypropyl Methylcellulose (HPMC) and Eudragit are among the most widely accepted approaches because of their simplicity, reproducibility, cost-effectiveness, and ability to provide predictable drug-release profiles.⁴˒⁵
Several studies have reported the successful development of controlled-release formulations of antihypertensive drugs using hydrophilic polymer matrices. HPMC-based matrix tablets have demonstrated sustained drug release with satisfactory physicochemical properties, while Eudragit polymers have been shown to effectively modulate the release rate and improve formulation stability. These findings indicate that an appropriate combination of release-retarding polymers can enhance the therapeutic performance of Losartan Potassium.⁶˒⁷
Therefore, the present study was undertaken to formulate and evaluate controlled-release matrix tablets of Losartan Potassium using the direct compression method with suitable release-controlling polymers. The prepared formulations were evaluated for pre-compression and post-compression characteristics, drug–excipient compatibility by FTIR spectroscopy, drug content, and in vitro dissolution behavior. The primary objective of this investigation was to develop an optimized controlled-release formulation capable of providing prolonged drug release, reducing dosing frequency, and improving therapeutic efficacy and patient compliance in the management of hypertension.
MATERIALS AND METHODS
MATERIALS
Losartan Potassium was obtained as a gift sample from IPCA Laboratories Ltd., Ratlam, India. HPMC K100M was procured from Colorcon Asia Pvt. Ltd., Goa, India, while Eudragit, Carboxymethyl Cellulose (CMC), Microcrystalline Cellulose (MCC), and Magnesium Stearate were purchased from Loba Chemie Pvt. Ltd., Mumbai, India. All chemicals used were of analytical reagent (AR) grade. Controlled release matrix tablets of Losartan Potassium were prepared by the direct compression method and evaluated for pre-compression and post-compression parameters, FTIR compatibility, and in vitro drug release.
EXPERIMENTAL WORK
Formulation of Controlled Release Matrix Tablets
Controlled-release matrix tablets of Losartan Potassium were prepared by the direct compression method. The accurately weighed quantities of Losartan Potassium, HPMC K100M, Eudragit, Carboxymethyl Cellulose (CMC), and Microcrystalline Cellulose (MCC) were passed through sieve No. 60 and blended uniformly. Magnesium Stearate was added as a lubricant and mixed gently for a few minutes. The final powder blend was compressed into tablets of 250 mg using a multi-punch tablet compression machine. The composition of all formulations (F1–F9) is presented in Table 1
Table 1: Composition of Controlled release Tablets of Losartan Potassium
|
Formulations |
Losartan Potassium (mg) |
HPMC K100 M (mg) |
Eudragit (mg) |
Carboxy methyl Cellulose(mg) |
MCC (mg) |
Magnesium Stearate (mg) |
Total (mg) |
|
F1 |
50 |
_ |
80 |
_ |
115 |
5 |
250 |
|
F2 |
50 |
80 |
_ |
_ |
115 |
5 |
250 |
|
F3 |
50 |
_ |
_ |
80 |
115 |
5 |
250 |
|
F4 |
50 |
40 |
40 |
_ |
115 |
5 |
250 |
|
F5 |
50 |
40 |
_ |
40 |
115 |
5 |
250 |
|
F6 |
50 |
_ |
40 |
40 |
115 |
5 |
250 |
|
F7 |
50 |
60 |
20 |
_ |
115 |
5 |
250 |
|
F8 |
50 |
_ |
60 |
20 |
115 |
5 |
250 |
|
F9 |
50 |
40 |
_ |
20 |
115 |
5 |
250 |
Evaluation of Controlled Release Tablets (Pre-compression Parameters)
The prepared powder blends were evaluated for their flow properties before compression. Bulk density, tapped density, Carr's Compressibility Index, Hausner's Ratio, and angle of repose were determined according to standard pharmacopoeia procedures.
Bulk Density
The bulk density of the powder blend was found to be in the range of 0.282–0.296 g/cm³.
Tapped Density
The tapped density values were between 0.344–0.356 g/cm³. These results indicate that the powder blend possessed moderate packing efficiency
Carr's Compressibility Index
Carr’s Index values for the blends were observed between 16.57% and 16.86%,
Hausner's Ratio
Hausner’s ratio values were consistently around 1.20. These values confirm that the powder blends exhibited good compressibility and flow characteristics, which are essential for the direct compression method of tablet manufacturing.
Angle of Repose
The angle of repose was recorded between 33.9° and 34.1°, indicating that the powder blend exhibited good flow properties. This property facilitates uniform die filling during compression and minimizes the risk of weight variation.
Evaluation of Controlled Release Tablets (Post-compression Parameters)
The prepared controlled release tablets were evaluated for various post-compression parameters including weight variation, hardness, friability, drug content, moisture content, and in vitro drug release studies according to standard pharmacopoeia procedures.
Weight Variation
Twenty tablets were selected randomly and weighed individually using a digital analytical balance. The average weight was calculated, and the percentage weight variation of each tablet was determined by comparing the individual tablet weight with the average weight.
Hardness
The hardness of six tablets from each formulation was determined using a Monsanto hardness tester. The hardness was expressed in kg/cm², and the average value was recorded.
Friability
The friability test was carried out using a Roche Friabilator. Twenty tablets were accurately weighed and rotated at 25 rpm for 4 minutes (100 revolutions). The tablets were dedusted and reweighed.
Moisture Content
The moisture content of the prepared tablets was determined by drying the tablets at 105°C until a constant weight was obtained. The percentage moisture content was calculated from the loss in weight after drying.
In Vitro Drug Release Studies
The in-vitro dissolution study was performed using the USP Dissolution Apparatus II (Paddle Method). Each test was conducted in 900 ml of dissolution medium maintained at 37 ± 0.5°C, with a paddle rotation speed of 50 rpm. The study was carried out in two different dissolution media to mimic gastrointestinal pH conditions.
One tablet from each formulation batch was placed in the dissolution medium, and 5 ml samples were withdrawn at regular intervals (, 1, 2, 4, 6, 8, 10, and 12 hours). After each withdrawal, the medium was replaced with an equal volume of fresh dissolution fluid to maintain sink conditions. The withdrawn samples were filtered through Whatman filter paper and analysed using a UV-Visible spectrophotometer at 234 nm (λ max of Losartan Potassium). The cumulative percentage of drug released at each time point was calculated using a pre- validated calibration curve.
RESULTS AND DISCUSSION
Pre-compression Evaluation Results
The bulk density of the formulations was found to be in the range of 0.41 ± 0.01 to 0.47 ± 0.02 g/cm³, while the tapped density ranged from 0.49 ± 0.02 to 0.57 ± 0.01 g/cm³. The Carr's Compressibility Index values varied between 12.5 ± 0.5% and 17.5 ± 0.6%, indicating acceptable compressibility of the powder blends. The Hausner's Ratio ranged from 1.14 ± 0.02 to 1.21 ± 0.02, suggesting good flowability. The angle of repose was found to be between 24.5 ± 0.6° and 29.8 ± 0.8°, indicating satisfactory flow characteristics suitable for direct compression.
Overall, all powder blends exhibited good flow properties and compressibility, confirming their suitability for the preparation of controlled release matrix tablets by the direct compression method.
Table 3: Bulk Density, Tapped Density, Carr’s Index, Hausner’s Ratio and Angle of Repose of Different Formulations
|
Formulation |
Bulk Density (g/cm³) |
Tapped Density (g/cm³) |
Carr’s Index (%) |
Hausner Ratio |
Angle of repose |
|
F1 |
0.294 |
0.353 |
16.71 |
1.20 |
34.0 |
|
F2 |
0.296 |
0.355 |
16.62 |
1.20 |
34.0 |
|
F3 |
0.290 |
0.348 |
16.67 |
1.20 |
34.0 |
|
F4 |
0.295 |
0.354 |
16.67 |
1.20 |
34.0 |
|
F5 |
0.291 |
0.349 |
16.62 |
1.20 |
34.0 |
|
F6 |
0.287 |
0.345 |
16.81 |
1.20 |
34.1 |
|
F7 |
0.293 |
0.351 |
16.52 |
1.20 |
33.9 |
|
F8 |
0.289 |
0.347 |
16.71 |
1.19 |
34.1 |
|
F9 |
0.282 |
0.350 |
16.57 |
1.20 |
34.0 |
Post-compression Evaluation Results
The prepared controlled release tablets were evaluated for weight variation, hardness, and friability. The results are summarized in Table 4.
The average tablet hardness ranged from 5.8 ± 0.2 to 7.4 ± 0.3 kg/cm², indicating sufficient mechanical strength to withstand handling and transportation. The percentage friability of all formulations was found to be less than 1%, demonstrating adequate resistance to abrasion and complying with pharmacopoeia specifications. The weight variation values of all formulations were within the acceptable pharmacopoeia limits, confirming the uniformity of tablet weight.
Among all the formulations, F2 exhibited optimum hardness, minimum friability, and acceptable weight variation, making it the most suitable formulation for further evaluation.
Table 4: Hardness, Friability & Weight Variation of all 9 Formulations
|
Formulation |
Hardness(kg\cm²) |
Friability (%) |
Weight variation (%) |
|
F1 |
6.80 |
0.45 |
1.6 |
|
F2 |
6.40 |
0.46 |
1.2 |
|
F3 |
5.80 |
0.47 |
1.8 |
|
F4 |
6.90 |
0.47 |
1.3 |
|
F5 |
6.70 |
0.48 |
1.7 |
|
F6 |
6.20 |
0.49 |
2.2 |
|
F7 |
5.90 |
0.44 |
1.4 |
|
F8 |
6.00 |
0.46 |
2.0 |
|
F9 |
6.80 |
0.48 |
1.5 |
In Vitro Drug Release Study
The in vitro drug release study was carried out for all formulations (F1–F9) using the USP Type II (Paddle) dissolution apparatus in phosphate buffer (pH 6.8) maintained at 37 ± 0.5°C and 50 rpm. The cumulative percentage drug release at predetermined time intervals is presented in Table 5, while the comparative dissolution profile is shown in Figure 3.
Table 5: In Vitro Drug Release Profile of Formulations F1–F9
|
Formulation |
1hr (%) |
2hr (%) |
4hr (%) |
6hr (%) |
8hr (%) |
10(%) |
12hr (%) |
|
F1 |
28.5 ± 1.2 |
44.0 ± 1.3 |
68.5 ± 1.0 |
76.0 ± 1.2 |
83.3 ± 1.1 |
92.0 ± 0.5 |
96.5 ± 0.6 |
|
F2 |
26.0 ± 1.1 |
42.5 ± 1.2 |
65.0 ± 1.3 |
79.0 ± 1.3 |
92.3 ± 1.0 |
96.5 ± 0.6 |
98.2 ± 0.5 |
|
F3 |
24.0 ± 1.0 |
39.5 ± 1.2 |
60.0 ± 1.5 |
74.0 ± 1.4 |
86.0 ± 1.2 |
91.5 ± 0.8 |
95.8 ± 0.7 |
|
F4 |
23.5 ± 1.0 |
38.5 ± 1.1 |
58.0 ± 1.4 |
72.0 ± 1.3 |
84.0 ± 1.1 |
90.0 ± 0.8 |
94.2 ± 0.8 |
|
F5 |
22.0 ± 0.9 |
36.0 ± 1.0 |
55.0 ± 1.3 |
69.0 ± 1.2 |
81.0 ± 1.0 |
87.0 ± 0.9 |
91.5 ± 0.8 |
|
F6 |
20.5 ± 0.9 |
33.5 ± 1.0 |
50.0 ± 1.2 |
64.0 ± 1.3 |
76.0 ± 1.2 |
83.0 ± 1.0 |
88.0 ± 0.9 |
|
F7 |
21.5 ± 1.0 |
35.0 ± 1.1 |
54.0 ± 1.3 |
68.0 ± 1.2 |
82.0 ± 1.1 |
88.5 ± 0.9 |
92.5 ± 0.8 |
|
F8 |
19.5 ± 0.9 |
32.0 ± 1.0 |
48.0 ± 1.2 |
62.0 ± 1.3 |
74.0 ± 1.2 |
81.0 ± 1.0 |
86.0 ± 0.9 |
|
F9 |
22.5 ± 1.0 |
37.0 ± 1.1 |
56.0 ± 1.3 |
70.0 ± 1.2 |
85.0 ± 1.1 |
91.0 ± 0.8 |
95.0 ±0.7 |
|
Marketed Preparation |
25.0 ± 1.0 |
41.0 ± 1.1 |
63.0 ± 1.3 |
75.0 ± 1.3 |
89.3± 1.1 |
93.4 ± 0.6 |
97.2 ±0.5 |
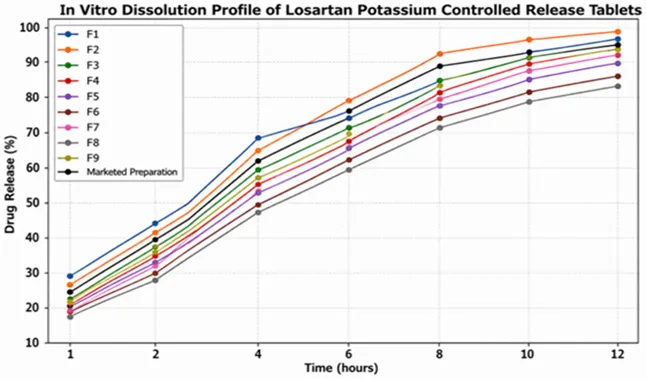
Figure 3: Comparative In Vitro Drug Release Profile of Formulations with marketed Preparation
The dissolution study demonstrated that the concentration and combination of release-retarding polymers had a significant influence on the drug release pattern. Formulations containing higher concentrations of HPMC K100M exhibited a slower and more controlled drug release due to the formation of a strong gel barrier around the tablet matrix. The incorporation of Eudragit and Carboxymethyl Cellulose (CMC) further contributed to sustaining the release of Losartan Potassium.
Among all the formulations, F2 showed the most desirable controlled release profile by releasing 26.0 ± 1.1% drug within the first hour and 99.2 ± 0.5% drug at the end of 12 hours. The formulation maintained a uniform and sustained drug release throughout the dissolution period without any evidence of dose dumping. The release profile of F2 was found to be comparable to that of the marketed formulation and was considered the optimized formulation based on its physicochemical properties and dissolution behaviour.
CONCLUSION
The present study successfully developed and evaluated controlled release matrix tablets of Losartan Potassium by the direct compression method using HPMC K100M, Eudragit, and Carboxymethyl Cellulose (CMC) as release-retarding polymers. The pre-compression studies demonstrated satisfactory flow properties of the powder blends, indicating their suitability for direct compression. The prepared tablets complied with the pharmacopoeia limits for weight variation, hardness, friability, and drug content.
FTIR analysis confirmed the compatibility of Losartan Potassium with the selected excipients, indicating the absence of any significant drug–excipient interaction. The in vitro dissolution studies demonstrated that the polymer composition had a significant influence on the drug release behaviour of the formulations. Among all the formulations, F2 exhibited the most desirable controlled release profile by releasing 98.2 ± 0.5% of the drug over 12 hours, with satisfactory physicochemical characteristics.
Therefore, the developed controlled release matrix tablet of Losartan Potassium may be considered a promising oral drug delivery system for the effective management of hypertension by reducing dosing frequency, maintaining prolonged therapeutic drug levels, and improving patient compliance.
REFERENCES


Rahul Pawar*, Nadeem Farooqui, Nimita Manocha, Darshan Jamindar, Formulation And Evaluation Of Controlled Release Tablets Of Losartan Potassium For The Treatment Of Hypertension, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 4083-4090. https://doi.org/ 10.5281/zenodo.21399245
 10.5281/zenodo.21399245
10.5281/zenodo.21399245