
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
MBBS Second Year Student, Microbiology, Virology and Immunology, Fergana Medical Institute of Public health, Uzbekistan
Background: Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains one of the foremost infectious causes of global mortality. Long-duration multi-drug regimens, while effective against drug-susceptible strains, are progressively undermined by the emergence of drug-resistant variants, including multidrug-resistant (MDR-TB), pre-extensively drug-resistant (Pre-XDR-TB), and extensively drug-resistant (XDR-TB) forms. Objective: This review consolidates current knowledge on the pharmacological mechanisms of first- and second-line anti-TB agents, the molecular pathways through which Mtb acquires intrinsic and adaptive resistance, and the consequent impact on clinical treatment outcomes. Methods: A structured narrative review was conducted using peer-reviewed literature sourced from PubMed, Scopus, and Web of Science databases, spanning publications up to 2026. Results: Resistance in Mtb arises predominantly through chromosomal point mutations in drug-target genes (rpoB, katG, inhA, pncA, embCAB, gyrA), augmented by efflux-pump overexpression, enzymatic drug inactivation, and cell-wall impermeability. These mechanisms collectively erode the efficacy of core agents including rifampicin, isoniazid, pyrazinamide, and ethambutol. Novel compounds—bedaquiline and delamanid—show substantial promise, although cross-resistance and pharmacokinetic challenges warrant vigilance. Diagnostic advances, including nucleic acid amplification tests (NAATs), line probe assays (LPAs), and whole-genome sequencing, markedly accelerate resistance detection. Treatment outcomes are adversely affected by comorbidities (diabetes mellitus, HIV, hepatic disease), advanced age, incomplete adherence, and pandemic-era disruptions. Conclusion: Addressing the TB drug-resistance crisis demands integrated strategies encompassing rational drug use, enhanced molecular diagnostics, host-directed therapies, nanoparticle-based delivery systems, and robust global surveillance frameworks.
INTRODUCTION
Tuberculosis ranks among the most consequential infectious diseases in recorded human history. Despite decades of international control efforts, Mtb continues to claim approximately 1.3 million lives annually, with projections forecasting tens of millions of additional deaths over the coming decades if current trends persist. The causative organism, Mycobacterium tuberculosis, is distinguished by its capacity to establish chronic, sometimes lifelong infection in host macrophages and to survive in a dormant state within pulmonary granulomas that are largely impervious to host immunity and antimicrobial agents alike.
Standard first-line therapy for drug-susceptible TB comprises a six-month regimen of isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA), and ethambutol (EMB)—a combination developed in the mid-twentieth century and still the therapeutic cornerstone today. While this regimen achieves high cure rates under optimal conditions, its protracted duration, demanding pill burden, and attendant toxicities create fertile ground for incomplete adherence. Sub-therapeutic drug exposure, in turn, generates selection pressure that progressively favours resistant bacterial sub-populations, driving the global emergence of MDR-TB—defined as resistance to at least INH and RIF—and the even more refractory XDR-TB phenotype.
The approval of two entirely new anti-TB agents—bedaquiline in 2012 and delamanid in 2014—represented the first substantive therapeutic addition in over four decades. Yet these agents alone cannot resolve the resistance crisis without a parallel revolution in rapid molecular diagnostics, rational prescribing, and novel delivery strategies. This review therefore examines, in an integrated manner, the pharmacological mechanisms of anti-TB drugs, the molecular architecture of Mtb resistance, the classification and clinical impact of drug-resistant disease phenotypes, contemporary diagnostic platforms, determinants of treatment outcome, management challenges, and emerging therapeutic strategies.
2. MECHANISMS OF ACTION OF ANTI-TB DRUGS
Anti-tuberculosis agents are conventionally stratified into first-line drugs—characterised by high bactericidal potency combined with an acceptable safety profile—and second-line drugs, which are reserved for resistant cases owing to comparatively lower efficacy or greater toxicity. Both classes employ diverse mechanisms, encompassing inhibition of cell-wall biosynthesis, disruption of nucleic acid metabolism, interference with protein synthesis, and impairment of membrane integrity.
2.1 First-Line Drugs
First-line agents form the backbone of initial TB therapy and are most effective against actively replicating bacilli during the early bactericidal phase of treatment.
2.1.1 Rifampicin
Rifampicin (RIF) is a lipophilic ansamycin antibiotic that exerts its bactericidal effect by forming a high-affinity binary complex with the beta (β) subunit of bacterial DNA-dependent RNA polymerase, encoded by the rpoB gene. With a binding constant of approximately 10⁻⁹ M at 37°C, the drug–enzyme complex potently blocks both RNA chain initiation and elongation. This inhibition arises from rifampicin’s steric interference with the binding sites for nascent RNA and the pyrophosphate reaction product within the enzyme’s catalytic pocket. Because rifampicin must traverse the bacterial cytoplasm to engage its target, mutants exhibiting reduced drug uptake across the cell membrane represent one pathway to phenotypic resistance.
2.1.2 Isoniazid
Isoniazid (INH) functions as a prodrug requiring activation by the mycobacterial catalase-peroxidase enzyme KatG. Upon activation, INH-derived reactive species—including isonicotinoyl radicals and nitric oxide—generate covalent adducts with the nicotinamide adenine dinucleotide (NAD⁺) and NADP⁺ cofactors. The resulting isonicotinoyl-NAD adduct is a potent competitive inhibitor of InhA, the NADH-dependent enoyl-acyl carrier protein (ACP) reductase central to the fatty acid synthase II system. Inhibition of InhA depletes mycolic acid precursors essential for cell-wall integrity, simultaneously exhausting cellular nucleic acid and lipid biosynthetic reserves. The concerted disruption of multiple anabolic pathways accounts for INH’s exceptional potency and selectivity against mycobacteria.
2.1.3 Pyrazinamide
Pyrazinamide (PZA) is an unusual prodrug that is passively taken up by tubercle bacilli and hydrolysed to pyrazinoic acid (POA) by the enzyme pyrazinamidase/nicotinamidase, encoded by the pncA gene. POA, under the acidic conditions encountered in inflammatory lesions and within macrophage phagolysosomes, becomes protonated and re-enters the bacterial cytoplasm by passive diffusion, where it accumulates to toxic concentrations. POA exerts pleiotropic bactericidal effects: disruption of membrane electrochemical potential, inhibition of trans-translation—a quality-control mechanism that rescues stalled ribosomes—and possible interference with coenzyme A biosynthesis. Strikingly, PZA activity increases as bacterial metabolic activity decreases, rendering it uniquely efficacious against non-replicating persisters that are refractory to most other agents and thereby enabling the shortening of total therapy duration.
2.1.4 Ethambutol
Ethambutol (EMB) is a bacteriostatic agent that selectively inhibits arabinosyl transferase enzymes encoded by the embCAB operon. These enzymes catalyse the polymerisation of arabinogalactan, a polysaccharide component that anchors mycolic acids to the peptidoglycan core of the mycobacterial cell wall. Disruption of arabinogalactan synthesis leads to accumulation of the intermediate D-arabinofuranosyl-P-decaprenol and progressive cell-wall destabilisation. Notably, EMB exhibits synergistic activity with INH by suppressing the inhA gene transcriptional repressor, thereby amplifying inhibition of the shared mycolic acid biosynthetic pathway.
2.2 Second-Line Drugs
Second-line agents are deployed when first-line regimens fail or are contraindicated, and are categorised by the World Health Organization (WHO) into Group A (fluoroquinolones), Group B (injectable aminoglycosides), and Group C (other core second-line agents including newer drugs).
2.2.1 Fluoroquinolones
Fluoroquinolones, encompassing moxifloxacin, levofloxacin, gatifloxacin, sparfloxacin, and ofloxacin, are broad-spectrum bactericidal agents with potent activity against Mtb. Their mechanism centres on the formation of ternary drug–DNA–enzyme complexes with DNA gyrase (the sole topoisomerase II in Mtb, encoded by gyrA and gyrB), thereby blocking the movement of DNA replication forks and transcription complexes. Because Mtb lacks topoisomerase IV—the secondary quinolone target in most other bacteria—mutations in the quinolone resistance-determining region (QRDR) of gyrA are the predominant mechanism of acquired fluoroquinolone resistance in mycobacteria. Newer-generation fluoroquinolones, particularly moxifloxacin, are under evaluation as potential first-line substitutes with the goal of shortening total treatment duration.
2.2.2 Aminoglycosides
Streptomycin, the first anti-TB drug discovered (1944), and the structurally related semi-synthetic derivatives kanamycin and amikacin belong to the aminoglycoside class. These agents bind irreversibly to the 30S ribosomal subunit of Mtb, inducing misreading of mRNA codons and premature termination of translation, ultimately disrupting cell-membrane integrity. Because aminoglycosides act preferentially on extracellular bacilli, their intracellular efficacy within macrophages is limited. Resistance arises chiefly through mutations in the rrs gene (encoding 16S ribosomal RNA) and the rpsL gene (encoding ribosomal protein S12).
2.2.3 Para-aminosalicylic Acid
Para-aminosalicylic acid (PAS), employed as a first-line agent from 1946 through the mid-1960s, is a bacteriostatic compound with a minimum inhibitory concentration (MIC) of approximately 1 μg/mL for Mtb. Its mechanism remains incompletely elucidated; however, current evidence implicates competitive inhibition of folate biosynthesis—by structural mimicry of para-aminobenzoic acid—and interference with mycobacterial iron acquisition pathways. PAS acts preferentially on extracellular organisms.
2.2.4 Cycloserine/Terizidone
Cycloserine, synthesised in 1952, is a structural analogue of D-alanine that competitively inhibits two enzymes essential for peptidoglycan biosynthesis: alanine racemase (which converts L-alanine to D-alanine) and D-alanyl-D-alanine synthetase (which forms the terminal dipeptide of the peptidoglycan precursor). Because these enzymes are indispensable for the rigidity and stability of the mycobacterial cell wall, their inhibition is lethal to proliferating bacteria. Terizidone, a condensation product of two cycloserine molecules, has equivalent activity and is associated with a somewhat more tolerable adverse-effect profile.
2.3 Summary of Molecular Targets and Mechanisms
Table 1. Molecular targets and mechanisms of principal anti-TB drugs.
|
Drug |
Class |
Primary Target |
Target Gene(s) |
Effect |
|
Rifampicin |
Ansamycin |
RNA polymerase β-subunit |
rpoB |
Blocks RNA synthesis |
|
Isoniazid |
Isonicotinic acid hydrazide |
InhA (enoyl-ACP reductase) |
katG, inhA |
Inhibits mycolic acid biosynthesis |
|
Pyrazinamide |
Pyrazine analogue |
PZase; membrane energy |
pncA |
Disrupts membrane potential |
|
Ethambutol |
Diamino-butanol |
Arabinosyl transferase |
embCAB |
Inhibits arabinogalactan synthesis |
|
Fluoroquinolones |
Quinolone |
DNA gyrase A/B |
gyrA, gyrB |
Blocks DNA replication |
|
Aminoglycosides |
Aminocyclitol |
30S ribosomal subunit |
rrs, rpsL |
Inhibits protein synthesis |
|
PAS |
Salicylate analogue |
Folate pathway; iron acquisition |
thyA, folC |
Bacteriostatic; depletes folate |
|
Cycloserine |
D-alanine analogue |
Alanine racemase; D-Ala-D-Ala synthetase |
alr, ddlA |
Disrupts peptidoglycan synthesis |
|
Bedaquiline |
Diarylquinoline |
ATP synthase (F₀ subunit) |
atpE |
Depletes cellular ATP |
|
Delamanid |
Nitroimidazo-oxazole |
Mycolic acid biosynthesis (F420-dependent) |
fbiA/B/C, ddn |
Blocks cell-wall lipid synthesis |
3. MOLECULAR MECHANISMS OF DRUG RESISTANCE IN M. TUBERCULOSIS
Drug resistance in Mtb is fundamentally a consequence of chromosomal mutation rather than horizontal gene transfer, which distinguishes the molecular epidemiology of TB from that of many other bacterial pathogens. Resistance may be intrinsic—arising from the organism’s inherent structural or biochemical properties—or acquired, driven by selection under drug pressure.
3.1 Intrinsic Resistance Mechanisms
The extraordinary impermeability of the mycobacterial cell wall constitutes the primary line of intrinsic defence. The outer leaflet of this wall is dominated by extremely long-chain, branched mycolic acids covalently anchored to an arabinogalactan–peptidoglycan scaffold, creating a hydrophobic barrier that retards the passive diffusion of hydrophilic antibiotics across the cell envelope. Complementing this physical barrier, Mtb elaborates an intrinsic β-lactamase (BlaC) that hydrolyses β-lactam antibiotics, and encodes the 23S rRNA methyltransferase erm(37), which methylates the macrolide binding site on the ribosome and confers natural resistance to this antibiotic class.
Beyond the cell wall, active drug efflux is a key determinant of intrinsic susceptibility. More than 30 distinct efflux pump systems spanning five major structural superfamilies have been identified in Mtb: the ATP-binding cassette (ABC) transporters, the resistance-nodulation-division (RND) family, the major facilitator superfamily (MFS), the small multidrug resistance (SMR) proteins, and the multidrug and toxic compound extrusion (MATE) family. Baseline expression of these transporters maintains sub-inhibitory intracellular drug concentrations. When pharmacological stress is applied, upregulation of pump expression—observable within hours of drug exposure—further reduces intracellular drug accumulation and effectively lowers the threshold for subsequent mutational resistance.
Additional intrinsic mechanisms include species-specific structural divergence of drug target sites (reducing drug binding affinity), enzymatic modification or degradation of antimicrobial molecules, and cell-cycle regulatory factors such as TapA (MSMEG_3748/Rv1697), which coordinates division and polar growth in mycobacteria. Loss of TapA function disrupts division, dysregulates envelope remodelling, and paradoxically sensitises Mtb to several first- and second-line drugs, suggesting that cell-cycle progression is intrinsically linked to antibiotic susceptibility phenotypes.

Fig.1 Mechanism of Intrinsic resistance in Mycobacterium tuberculosis
3.2 Acquired Resistance Mechanisms
Acquired drug resistance in Mtb emerges through the selection of pre-existing spontaneous chromosomal mutations that confer survival under drug pressure. The mutation rate in Mtb is estimated at approximately 2–8 × 10⁻¹⁰ per base pair per replication cycle, and in the large bacterial populations present in cavitary pulmonary disease, resistant mutants arise stochastically. Monotherapy, sub-therapeutic dosing, and irregular adherence create the selective milieu for their fixation and eventual dominance.
3.2.1 Rifampicin Resistance
Approximately 95% of rifampicin-resistant Mtb isolates carry mutations within an 81-base-pair rifampicin resistance-determining region (RRDR) of the rpoB gene. Codons 531 (Ser→Leu), 526 (His→Asp/Tyr), and 516 (Asp→Val) account for the majority of substitutions observed globally. Codon 531 mutations produce complete loss of rifampicin binding affinity, while codon 526 mutations are associated with cross-resistance to the rifamycin class. Rare mutations outside the RRDR account for the minority of phenotypically resistant isolates in whom rpoB RRDR screening is falsely negative.
3.2.2 Isoniazid Resistance
Resistance to INH arises through multiple independent mutational routes, reflecting the drug’s multifactorial mechanism. Mutations in katG—most commonly the Ser315Thr substitution—account for 50–70% of INH-resistant isolates and abolish prodrug activation. Mutations in the inhA promoter region or the inhA structural gene itself—by overexpressing or structurally altering the drug target—account for an additional 15–20% of cases and are associated with lower-level resistance. Concurrent mutations in the oxyR-ahpC intergenic region, typically in conjunction with katG mutations, contribute to the remaining resistant fraction. The heterogeneous genetic basis of INH resistance complicates both phenotypic and genotypic diagnostics.
3.2.3 Pyrazinamide Resistance
Resistance to PZA most commonly results from loss-of-function mutations dispersed throughout the pncA gene, which abolish PZase enzyme activity and prevent bioactivation of the prodrug. Over 300 distinct pncA mutations conferring resistance have been catalogued. Because no single mutational hotspot dominates, molecular detection of PZA resistance requires comprehensive pncA sequencing rather than targeted probing—a practical limitation in resource-constrained settings. Less common mechanisms include mutations affecting the bacterial efflux pump that extrudes POA before toxic intracellular concentrations are achieved.
3.2.4 Ethambutol Resistance
EMB resistance is primarily conferred by missense mutations in embB (most frequently at codon 306, Met→Ile/Val/Leu/Thr), encoding the catalytic subunit of arabinosyl transferase. Additional mutations in embA and embC, as well as upstream regulatory mutations that overexpress the wild-type embCAB operon, contribute to the resistance phenotype. The sensitivity of molecular tests for EMB resistance is lower than for rifampicin, because embB mutations alone do not always predict phenotypic resistance reliably.
3.2.5 Fluoroquinolone Resistance
Mutations in the QRDR of gyrA—particularly at codons 90 (Ala→Val), 91 (Ser→Pro), and 94 (Asp→Gly/His/Asn)—are the predominant drivers of fluoroquinolone resistance in Mtb, and their presence is a diagnostic criterion for pre-XDR-TB classification. GyrB mutations at codons 485, 500, and 543 contribute to lower-level resistance. Overexpression of efflux pumps—including MmpL5 and Rv1634—confers an additional layer of low-level resistance that may facilitate stepwise QRDR mutation acquisition under fluoroquinolone exposure.
3.2.6 Efflux-Mediated Resistance
Efflux pump upregulation constitutes a rapid, pre-mutational adaptive response that reduces effective intracellular drug concentrations across multiple drug classes simultaneously. The Mmr pump is significantly overexpressed in strains exposed to INH. The ABC-type pump Rv1819c has been recently structurally characterised and shown to confer resistance to multiple drug classes. Natural products including carvacrol, nobiletin, and carvotacetones, and synthetic compounds including verapamil analogues and 1,4-dihydropyridines, have demonstrated efflux pump inhibitory (EPI) activity, offering the conceptual basis for EPI adjuvant therapy to restore drug susceptibility.
3.3 Bedaquiline and Delamanid Resistance
Bedaquiline (BDQ) inhibits mycobacterial ATP synthase by binding the c-subunit (atpE) of the F₀ rotor, thereby collapsing the proton motive force and depleting cellular ATP. Resistance arises through atpE mutations that reduce drug binding, and through upregulation of the MmpS5-MmpL5 efflux system, which also mediates cross-resistance between BDQ and clofazimine (CFZ). Delamanid, a nitroimidazo-oxazole prodrug, requires reductive activation by the F420 deazaflavin-dependent nitroreductase Ddn; mutations in ddn or in the F420 biosynthetic genes (fbiA, fbiB, fbiC) abolish activation and confer resistance. The dual threat of phenotypic cross-resistance between BDQ and CFZ and co-resistance with established second-line agents underscores the importance of drug susceptibility testing (DST) before deploying these newer agents.
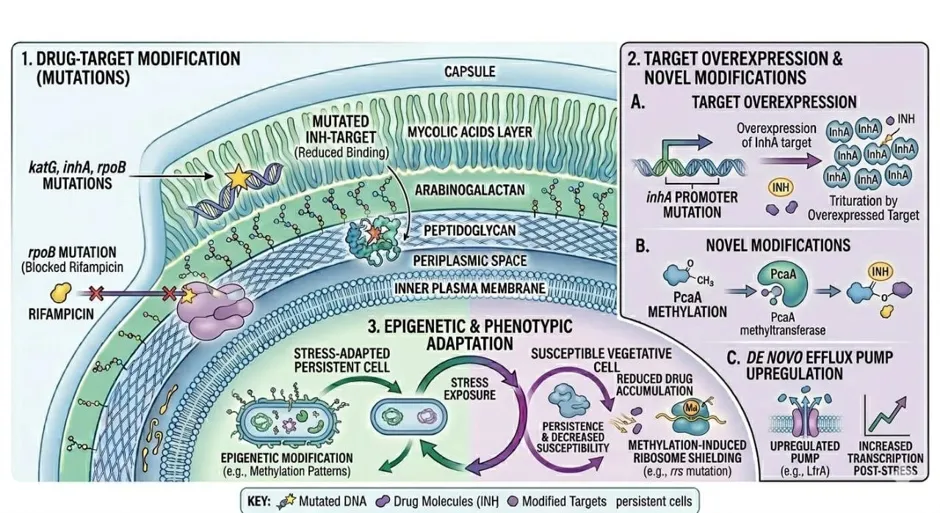
Fig.2 Mechanism of Extrinsic resistance in Mycobacterium tuberculosis
4. CLASSIFICATION OF DRUG-RESISTANT TUBERCULOSIS
The WHO recognises a hierarchical classification of drug-resistant TB phenotypes based on the pattern and breadth of resistance, each carrying distinct therapeutic implications:
Table 2. WHO classification of drug-resistant tuberculosis phenotypes and clinical implications.
|
Classification |
Resistance Profile |
Clinical Implication |
|
Mono-resistant TB |
Resistant to one first-line drug only (INH or RIF) |
May progress to MDR-TB if untreated |
|
Hr-TB |
INH-resistant, RIF-susceptible |
Most prevalent DR-TB form globally |
|
RR-TB |
RIF-resistant (with or without INH resistance) |
Indicator of potential MDR-TB |
|
MDR-TB |
Resistant to INH + RIF (minimum) |
Requires second-line therapy (18-24 months) |
|
Pre-XDR-TB |
MDR-TB + resistance to any fluoroquinolone |
Precursor to XDR-TB; limits regimen options |
|
XDR-TB |
Pre-XDR + resistance to BDQ and/or linezolid |
Very limited treatment options; poor outcomes |
|
TDR-TB |
Resistant to all tested first- and second-line drugs |
No established standard regimen; experimental therapy |
4.1 Factors Contributing to Resistance Emergence
The dynamics of resistance emergence in Mtb populations are governed by an interplay of clonal interference, variable mutation rates across genomic loci, efflux pump activity, compensatory mutations that restore bacterial fitness after resistance mutations reduce it, and epistatic interactions between mutations. Exposure to anti-TB drugs induces a bacterial stress response within hours, rapidly upregulating efflux pumps and generating a low-level resistance phenotype that, while insufficient for clinical resistance alone, provides sufficient selective advantage for bacillary survival and replication under sub-therapeutic drug concentrations. Subsequent accumulation of classical target-gene mutations then establishes stable, high-level, heritable resistance.
Within lung microenvironments, drug-resistant Mtb adapts further through modulation of cell-envelope lipid composition. Altered levels of free fatty acids, trehalose dimycolate (TDM), phthiocerol dimycocerosates (PDIMs), and phenolic glycolipids (PGLs) modify the surface properties of drug-resistant bacilli in ways that influence both host immune recognition and antibiotic penetration. During cavitary disease, extracellular drug-resistant Mtb may additionally secrete lipids that constitute an extracellular matrix-like shield further protecting the organism from antibiotic exposure.
5. DIAGNOSTIC APPROACHES FOR TUBERCULOSIS
Timely and accurate TB diagnosis is a prerequisite for effective disease management, transmission interruption, and resistance surveillance. The ideal diagnostic tool must combine sensitivity and specificity with rapidity, affordability, and operational simplicity sufficient for deployment in low-resource, high-burden settings. Current diagnostic approaches span microbiological, molecular, immunological, and emerging aptamer-based technologies.
5.1 Sputum Smear Microscopy
Sputum smear microscopy (SSM) remains the most widely deployed TB diagnostic tool, particularly in peripheral health facilities across high-burden countries. Two staining methods are principally employed: Ziehl-Neelsen (ZN) carbol-fuchsin staining, which produces red acid-fast bacilli against a blue methylene background; and fluorescence microscopy using auramine-O or auramine-rhodamine dyes, which renders bacilli fluorescent yellow-green against a dark field. While SSM is inexpensive (mean cost approximately USD 13 per test) and rapid, its sensitivity is fundamentally constrained by bacterial load, requiring a minimum of 10,000 bacilli per millilitre of sputum for reliable positivity. Sensitivity ranges from 32–89% depending on setting, specimen quality, and reader expertise. Critically, SSM cannot distinguish between live and dead organisms, drug-susceptible from drug-resistant strains, or Mtb from non-tuberculous mycobacteria (NTM).
5.2 Mycobacterial Culture
Liquid culture systems, notably the Mycobacterial Growth Indicator Tube (MGIT) platform, represent the gold standard for TB diagnosis, offering sensitivity approaching 100% for smear-negative disease and enabling subsequent drug susceptibility testing (DST) of recovered isolates. However, culture requires Biosafety Level 3 laboratory infrastructure due to the transmissibility of Mtb, yields results only after 2–6 weeks of incubation, and is prohibitively expensive for many endemic countries. False-positive culture rates of 1.6–4.7% further complicate interpretation in low-prevalence settings.
5.3 Molecular Diagnostic Platforms
5.3.1 Nucleic Acid Amplification Tests (NAATs)
NAATs, exemplified by the WHO-endorsed Xpert MTB/RIF and the newer Xpert MTB/RIF Ultra assays, detect Mtb-specific DNA sequences directly from unprocessed sputum specimens within 1–2 hours. Xpert MTB/RIF simultaneously screens for the most prevalent rpoB mutations, providing rifampicin resistance results concurrently with TB diagnosis. Sensitivity reaches 88–90% in smear-positive specimens and 61–76% in smear-negative specimens. These platforms have substantially expanded diagnostic access in primary care settings, though their cost and cartridge supply-chain requirements remain barriers in the most resource-constrained environments.
5.3.2 Line Probe Assays (LPAs)
LPAs employ PCR amplification followed by reverse-hybridisation of labelled amplicons to nitrocellulose membrane-bound oligonucleotide probes targeting specific resistance-conferring mutations. The GenoType MTBDRplus assay detects rpoB (RRDR), katG (codon 315), and inhA (promoter region) mutations, covering >95% of RIF-resistant and ~80–90% of INH-resistant strains. The expanded GenoType MTBDRsl assay extends coverage to second-line resistance, interrogating gyrA, gyrB, rrs, and eis mutations relevant to fluoroquinolone and aminoglycoside resistance. LPAs yield results within one working day but require intermediate laboratory capacity, including PCR infrastructure and contamination control.
5.3.3 Whole-Genome Sequencing (WGS)
WGS represents the most comprehensive tool for molecular epidemiology, phylogenetic typing, and resistance prediction, offering simultaneous interrogation of the entire Mtb genome to identify known and novel resistance mutations, transmission clusters, and mixed infections. While historically confined to reference laboratories, decreasing costs and improved bioinformatic pipelines are advancing WGS toward broader clinical application. Its limitations include turnaround times of several days, complex data interpretation requirements, and the ongoing challenge of correlating novel mutations with phenotypic resistance.
5.4 Immunological Diagnostic Approaches
Interferon-gamma release assays (IGRAs)—including the QuantiFERON-TB Gold Plus ELISA-based assay and the T-SPOT.TB ELISPOT-based assay—detect antigen-specific T-cell immune responses to the Mtb-specific proteins ESAT-6 and CFP-10. IGRAs have superior specificity compared to the tuberculin skin test (TST/Mantoux test) because ESAT-6 and CFP-10 are absent from BCG vaccine strains and most NTM, eliminating false-positive cross-reactions. However, neither IGRAs nor TST can reliably distinguish latent TB infection (LTBI) from active TB disease, nor predict progression risk. Novel Mtb antigen-based skin tests (TBSTs)—including C-Tb, Diaskintest, and the ESAT6-CFP10 test—introduced since 2022, aim to combine the accessibility of intradermal testing with the specificity advantages of IGRA antigens, with reported sensitivity and specificity exceeding 90% for Diaskintest.
6. IMPACT ON TREATMENT OUTCOMES
Treatment outcome in TB is assessed according to WHO-standardised definitions: cure (completion of therapy with bacteriological confirmation of clearance), treatment completion (without final bacteriological confirmation), treatment failure (smear-positive at five months despite supervised therapy), default/loss to follow-up (treatment interruption for two or more consecutive months), death during therapy, and transfer-out (outcome unknown). Successful treatment incorporates cured and completed categories; unsuccessful outcomes encompass failure, default, and death.
6.1 Drug Resistance and Treatment Success
Drug resistance profoundly impairs treatment success rates. For drug-susceptible TB, first-line therapy achieves success rates of 85–95% under directly observed conditions. MDR-TB success rates historically averaged 48% globally under long-course regimens, while XDR-TB was associated with success rates as low as 22–28% and mortality rates of 44–49%. The introduction of the all-oral BPaLM regimen (bedaquiline, pretomanid, linezolid, moxifloxacin) in the 2022 WHO treatment guidelines update represents a paradigm shift, offering substantially improved outcomes—with cure rates exceeding 89% in the landmark ZeNix and TB-PRACTECAL trials—in a condensed six-month duration.
6.2 Comorbidities and Adverse Outcomes
Comorbidities exert a profound adverse influence on TB treatment outcomes, both by compromising host immunity and by complicating drug pharmacokinetics. In a large multi-centre retrospective cohort of 1355 RR-TB patients in China, 49.5% carried at least one comorbidity. Diabetes mellitus was the most prevalent (17.8%), followed by other respiratory diseases (11.1%), hypertension (5.2%), immunodeficiency (4.9%), and viral hepatitis (3.9%). In multivariate analysis, diabetes (relative risk [RR] 1.31), severe heart disease (RR 1.70), malignancy (RR 1.89), and age 45 years or older (RR 1.38–1.64) all significantly increased the risk of unfavourable outcomes. Conversely, higher educational attainment, employment, and normal or overweight body mass index were protective.
HIV coinfection multiplicatively amplifies TB-related morbidity and mortality, with HIV-positive individuals experiencing an estimated 40-fold greater disability-adjusted life years (DALYs) from RR-TB compared to HIV-uninfected individuals. Rapid progression to disseminated or meningeal disease in the context of severe immunosuppression further worsens prognosis. Integration of antiretroviral therapy, particularly regimens incorporating the integrase inhibitor dolutegravir, with anti-TB therapy has simplified management and reduced serious adverse events in co-infected populations.
Diabetes mellitus independently increases susceptibility to primary TB infection and active disease, delays sputum culture conversion under treatment, and raises the risk of relapse after therapy completion. Optimal glycaemic control—defined as HbA1c below 7%—has been shown in systematic review and meta-analysis to significantly reduce complication rates and improve treatment outcomes in TB–DM co-morbid patients, underscoring the value of integrated chronic disease management in TB care pathways.
6.3 COVID-19 Pandemic Disruptions
The COVID-19 pandemic substantially disrupted TB control programmes globally between 2020 and 2022. Reallocation of healthcare resources toward pandemic management led to decreased TB case notifications, delayed diagnosis, and reduced treatment enrolment—conditions collectively favourable for transmission of drug-resistant strains. A meta-analysis of European epidemiological data demonstrated a simultaneous decrease in total TB notifications alongside an increase in the proportional representation of MDR and RR/MDR cases, while 24-month treatment success rates modestly improved as shorter regimens declined. Long-term effects on global resistance burden are anticipated to extend well beyond 2025, necessitating accelerated investment in TB diagnostic infrastructure and treatment access.
6.4 Pharmacokinetic Determinants of Outcome
Drug exposure, as measured by the area under the drug concentration–time curve (AUC) relative to the minimum inhibitory concentration (MIC), is a critical pharmacodynamic driver of treatment response in MDR-TB. In a multicentre prospective cohort study, patients achieving fluoroquinolone AUC₀₋₂₄/MIC above 231 (for moxifloxacin) and linezolid AUC₀₋₂₄/MIC above 287 had significantly higher rates of two-month culture conversion and favourable six-month outcomes. The probability of target attainment was highly variable across drugs—from 0% for ethambutol to 97% for linezolid—highlighting the need for therapeutic drug monitoring (TDM) to individualise dosing in complex MDR-TB patients.
7. CHALLENGES IN TUBERCULOSIS MANAGEMENT
7.1 Treatment Adherence and DOTS Implementation
Non-adherence to prolonged anti-TB regimens remains the principal modifiable driver of acquired drug resistance. The WHO directly observed treatment, short course (DOTS) strategy mandates supervised medication ingestion to ensure treatment completion. However, DOTS implementation in resource-limited settings is impeded by healthcare worker shortages, geographic inaccessibility, patient mobility, stigma, and economic barriers to clinic attendance. Digital adherence technologies, including electronic medication monitoring devices, video-observed therapy (VOT), and electronic patient service systems, have demonstrated improvements in first clinic visit compliance and monthly medication adherence rates, offering scalable adjuncts to conventional DOTS.
7.2 TB-HIV Integration
Despite evidence demonstrating that integrated TB–HIV service delivery reduces mortality and morbidity attributable to both conditions, implementation remains suboptimal in many high-burden settings, particularly in sub-Saharan Africa. Key advances enabling improved management include scale-up of urine-based lateral flow lipoarabinomannan (LF-LAM) assay for rapid TB screening in people living with HIV (PLWH), expanded access to bedaquiline and other newer DR-TB drugs, and dolutegravir-based antiretroviral regimens that minimise pharmacokinetic interactions with rifampicin. Cigarette smoking, prevalent in settings with high TB–HIV co-burden, further compounds immunological susceptibility to both infections and represents an under addressed modifiable risk factor.
7.3 Drug Toxicity and Adverse Reactions
Both first- and second-line anti-TB drugs carry significant adverse effect burdens that contribute to treatment interruption and non-completion. First-line drug toxicities include hepatotoxicity (INH, RIF, PZA), peripheral neuropathy (INH, cycloserine), optic neuritis (EMB), and hyperuricaemia (PZA). Among second-line agents, aminoglycosides carry risks of irreversible ototoxicity and nephrotoxicity; fluoroquinolones may prolong the QTc interval; cycloserine commonly causes neuropsychiatric adverse effects including psychosis and seizures; and ethionamide/ prothionamide produce hypothyroidism and severe gastrointestinal intolerance. Bedaquiline and delamanid both prolong QTc, necessitating baseline and serial electrocardiographic monitoring. In elderly patients, advanced age compounds these risks, with adverse drug reaction (ADR) incidence reaching 56.8% in patients over 80 years, predominantly gastrointestinal, neurological, and dermatological in nature.
7.4 Programmatic Management of TB Infection (TBI)
Prevention of progression from latent TB infection to active disease through TB preventive therapy (TPT) is an integral component of the WHO End-TB Strategy. However, programmatic implementation of TBI management is hampered by limitations in diagnostic tools to confirm infection (neither TST nor IGRAs achieve the required sensitivity-specificity profile in all clinical contexts), operational challenges in drug supply and healthcare worker training, and community reluctance toward preventive treatment. Among PLWH, PLHIV children aged under five years as household contacts, and migrants from high-incidence countries, the WHO recommends initiating TPT without mandatory confirmation of TBI in resource-limited settings.
8. NOVEL THERAPEUTIC STRATEGIES
8.1 Shortened Treatment Regimens
A four-month combination regimen comprising rifapentine, isoniazid, moxifloxacin, and pyrazinamide (HPMZ) has demonstrated non-inferiority to the standard six-month RIPE regimen for drug-susceptible pulmonary TB in the pivotal Study 31/ACTG A5349 randomised trial, representing the first successful shortening of first-line TB therapy duration in decades. Eligibility is currently restricted to adults and adolescents over 12 years, weight above 40 kg, HIV-positive patients with CD4 count above 100 cells/mm³, and patients with pulmonary (not severe extrapulmonary) disease. The substantially higher daily pill count compared to RIPE (13–15 vs. 7–12 pills) presents an adherence challenge requiring patient counselling and pharmacist support.
8.2 Host-Directed Therapies (HDT)
Host-directed therapies represent a conceptually distinct approach to TB treatment, targeting host immune pathways rather than the bacterium itself. By augmenting the host’s capacity to recognise, contain, and eliminate Mtb, HDTs aim to improve therapeutic outcomes, shorten treatment duration, and reduce tissue-destructive immunopathology. Among emerging HDT candidates, sulphasalazine inhibits the xCT amino acid transporter, reducing intracellular bacterial burden and attenuating pulmonary lesion severity in preclinical models. Imatinib, a tyrosine kinase inhibitor approved for chronic myeloid leukaemia, promotes phagosome maturation and enhances macrophage killing capacity. Vitamin D3 and sodium butyrate induce autophagy, facilitating lysosomal degradation of intracellular Mtb. These agents are currently being evaluated in clinical trials as adjuncts to standard anti-TB chemotherapy.
8.3 Nanoparticle-Based Drug Delivery Systems
Nanoparticle (NP) drug delivery systems offer several theoretical advantages in TB treatment: selective targeting of alveolar macrophages (the primary intracellular niche of Mtb), improved intracellular drug concentrations, extended drug release kinetics reducing dosing frequency, and the potential to co-encapsulate complementary drugs for synergistic effects. Particle sizes of 50–200 nm are optimal for phagocytic uptake while evading mucociliary clearance. By bypassing first-pass hepatic metabolism and reducing systemic drug exposure, NP systems may additionally reduce systemic toxicity and improve tolerability of agents such as INH and rifampicin.
Silver nanoparticles (AgNPs) and other metallic nanoparticles have demonstrated intrinsic antimycobacterial activity in preclinical studies. Unlike conventional antibiotics, metallic nanoparticles appear to operate through multiple simultaneous mechanisms—membrane disruption, oxidative stress generation, and interference with bacterial enzyme systems—making the development of stable resistance substantially more difficult. Nanoparticle-based aerosol vaccine platforms are also under development, offering a potential new modality for mucosal immunisation against TB.
8.4 New Drug Development
The current TB drug development pipeline includes compounds targeting novel Mtb vulnerabilities not exploited by existing agents. Inhibitors of DNA replication and repair enzymes, agents targeting mycobacterial energy metabolism beyond ATP synthase, and compounds that disrupt Mtb transcriptional regulation during latency represent active investigational areas. Third-line salvage options including linezolid, rifabutin, and repurposed agents such as thioridazine are being evaluated in compassionate-use and clinical trial contexts for TDR-TB. Prudent deployment of newly approved drugs in combination regimens—to minimise monotherapy-equivalent exposure—remains essential to preserving their long-term clinical utility.
8.5 Vaccination Strategies
BCG, the only licensed TB vaccine, provides good protection against severe childhood forms of TB (tuberculous meningitis, miliary TB) but inconsistent efficacy against adult pulmonary TB—the epidemiologically dominant form. Several novel vaccine candidates are progressing through clinical evaluation: M72/AS01E (a recombinant protein subunit vaccine that demonstrated approximately 50% efficacy against pulmonary TB in phase 2b trials); VPM1002 (a recombinant BCG strain); and MTBVAC (a live-attenuated Mtb-derived vaccine). Phase 3 efficacy trials, required for regulatory approval, are ongoing. Nanoparticle-based mucosal vaccine platforms represent a future investigational frontier aimed at establishing respiratory tract-localised immunity at the primary infection site.
9. CONCLUSION
Tuberculosis remains a formidable global health challenge, rendered increasingly complex by the inexorable emergence and spread of drug-resistant Mycobacterium tuberculosis strains. The molecular architecture of Mtb resistance—governed by chromosomal mutations in drug-target genes, augmented by efflux pump upregulation, enzymatic drug inactivation, and the extraordinary impermeability of the mycobacterial cell wall—creates a multi-layered defence against pharmacological intervention that demands a correspondingly multi-layered therapeutic response.
Advances in molecular diagnostics, particularly NAATs, LPAs, and whole-genome sequencing, have substantially accelerated the detection of resistance and enabled more rational regimen design. The approval of bedaquiline and delamanid, and the validation of the BPaLM regimen for MDR/XDR-TB, represent genuine therapeutic milestones. Simultaneously, the HPMZ four-month regimen offers hope for shortening the duration of treatment for drug-susceptible TB. Emerging strategies encompassing host-directed therapies, nanoparticle drug delivery, and next-generation vaccines broaden the therapeutic repertoire beyond conventional antimicrobial pharmacology.
Despite these advances, critical gaps persist. The majority of individuals with drug-resistant TB still lack access to effective treatment. Comorbidities—particularly HIV, diabetes mellitus, and malnutrition—adversely modulate treatment outcomes and demand integrated, patient-centred care pathways. The COVID-19 pandemic demonstrated the fragility of TB control programmes under system-level stress, with rebound effects on resistance trends that will persist for years. Achieving the WHO End-TB Strategy targets requires sustained multidisciplinary investment encompassing laboratory capacity building, new drug discovery, regulatory reform, and the political will to prioritise tuberculosis elimination in global health agendas.
REFERENCES

Padmalalitha Lakshmanan, Shamini Pushpakumari Santhoshkumar, Nisfa Mansuri Latifbhai, Dr. Abdul Latif, Molecular Basis of Antibiotic Resistance in Mycobacterium tuberculosis and its Impact on Treatment Outcomes: A Comprehensive Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 2772-2788. https://doi.org/10.5281/zenodo.20629434
 10.5281/zenodo.20629434
10.5281/zenodo.20629434