
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, College of Pharmaceutical Sciences, Government Medical College, Thiruvananthapuram
Thiazolidinedione derivatives are an important class of heterocyclic compounds primarily known for their antidiabetic activity through modulation of peroxisome proliferator-activated receptor gamma (PPAR-?). Recent studies have demonstrated their broader pharmacological potential in cancer and inflammatory disorders by influencing apoptosis, angiogenesis, oxidative stress, cell proliferation, and inflammatory pathways. The strong association between diabetes, chronic inflammation, and cancer progression has increased interest in multi-target therapeutic strategies. In this regard, thiazolidinedione-based hybrid molecules have emerged as promising candidates by combining the pharmacological benefits of the thiazolidinedione scaffold with other bioactive moieties to improve efficacy and selectivity. This review discusses recent advances in the design and development of TZD-based hybrids with enhanced therapeutic potential against diabetes, cancer, and inflammatory disorders. Various TZD derivatives have shown promising biological activities through multitarget actions such as regulation of glucose metabolism, inhibition of cancer cell growth, induction of apoptosis, and suppression of inflammatory pathways. This review also highlights the importance of molecular hybridization and computational approaches in developing safer, more selective, and effective TZD-based therapeutics for the management of diabetes-associated cancer and inflammation.
Cancer and diabetes mellitus are major global health challenges with significant morbidity and mortality worldwide. Chronic inflammation acts as a common pathological link between diabetes and cancer progression, contributing to oxidative stress, angiogenesis, and cellular proliferation. Cancer is characterized by uncontrolled cell proliferation, invasion, and metastasis, and remains one of the leading causes of death globally1. Diabetes mellitus, particularly Type 2 diabetes (T2DM), is a chronic metabolic disorder associated with persistent hyperglycemia, insulin resistance, and often hyperinsulinemia, and is associated with long-term complications, including nephropathy, neuropathy, retinopathy, and cardiovascular disorders2.
In T2DM, prolonged hyperglycemia and insulin resistance promote cellular damage through glucotoxicity, lipotoxicity, oxidative stress, and activation of inflammatory pathways such as nuclear factor-kappa B (NF-κB), phosphoinositide 3-kinase/protein kinase B (PI3K/Akt), and signal transducer and activator of transcription 3 (STAT3)3. These molecular alterations result in increased secretion of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β), thereby creating a pro-tumorigenic microenvironment that supports abnormal cell proliferation, angiogenesis, invasion, and metastasis, ultimately increasing cancer risk3,4. Epidemiological evidence suggests that T2DM increases the risk of several malignancies, including liver, pancreatic, colorectal, breast, endometrial, and bladder cancers, highlighting a strong pathological link between metabolic disorders and tumor progression1,4.
Thiazolidinediones (TZDs) or glitazones represent an important class of insulin sensitisers used in the management of T2DM, primarily by activating peroxisome proliferator-activated receptor gamma (PPAR-γ), a nuclear receptor involved in glucose and lipid metabolism5. Recently, activation of PPAR-γ by TZDs has been associated with significant anticancer and anti-inflammatory effects through regulation of cell differentiation, inhibition of cellular proliferation, induction of cell cycle arrest and apoptosis, suppression of tumor angiogenesis, and reduction of inflammatory mediators and pro-inflammatory cytokines6-8.
TZD scaffold can be substituted at the 3rd and 5th positions with different moieties to increase its potential activity9. 2,4-TZD core is often combined with diverse heterocyclic substitutes, resulting in the generation of distinct lead compounds which are investigated for their potential to act as antidiabetic, anticancer, anti-inflammatory, and antioxidant agents10-13. Therefore, thiazolidinedione-based derivatives and molecular hybrids have emerged as promising multi-target therapeutic agents for managing diabetes-associated cancer and inflammation by simultaneously modulating metabolic, proliferative, and inflammatory pathways.
2. TZDs as multitarget therapeutic agents
2.1 TZDs as antidiabetic agents
The antidiabetic activity of TZDs is primarily mediated through activation of PPAR-γ, a ligand-activated nuclear receptor involved in the regulation of glucose and lipid metabolism, and insulin sensitivity5,14,15. Upon activation, PPAR-γ forms a heterodimer with the retinoid X receptor (RXR) and binds to peroxisome proliferator response elements (PPREs), leading to modulation of genes involved in glucose homeostasis and lipid utilization16-18. Activation of PPAR-γ by TZDs enhances peripheral glucose uptake, improves insulin sensitivity, reduces hepatic gluconeogenesis, and suppresses inflammatory mediators, thereby improving glycemic control in diabetic patients16,19. Although clinically approved TZDs such as pioglitazone and rosiglitazone are effective antidiabetic agents, their associated adverse effects have prompted the development of novel TZD derivatives with improved efficacy and safety profiles18,20. Consequently, structural modifications of the TZD scaffold have been extensively explored to design potent PPAR-γ modulators with enhanced antidiabetic potential20,21.
Pioglitazone Rosiglitazone
Somayeh Behrouz et al. designed and synthesized a series of novel 5-arylidene thiazolidinedione acetic acid 1,2-diol monoesters as potential antidiabetic agents. The synthesized compounds were characterized using various spectroscopic techniques and evaluated for their antidiabetic potential in streptozotocin-induced diabetic male Balb/C mice over four weeks. Most derivatives exhibited significant hypoglycemic activity compared to glibenclamide. Among them, compound 2c showed the most promising blood glucose-lowering effect without mortality. Molecular docking studies demonstrated a strong binding affinity of compound 2c towards the active site of the PPAR-γ receptor, supporting its mechanism as a potential PPAR-γ modulator. In silico ADMET and physicochemical studies of compounds 2a–2p revealed favorable drug-likeness and pharmacokinetic profiles. DFT analysis further demonstrated similarities between compound 2c and rosiglitazone, highlighting 2c as a potential lead candidate for diabetes therapy21. Mahendra Gowdru Srinivasa et al. synthesized and characterized a series of novel thiazolidinedione derivatives and evaluated their antidiabetic potential as PPAR-γ agonists. Molecular docking, MM-GBSA, and 200 ns molecular dynamics simulations demonstrated strong and stable binding interactions of compounds 3d, 3g, and 3h with the PPAR-γ receptor, comparable to pioglitazone. ADME analysis indicated favorable drug-likeness properties. In vitro studies, including MTT cytotoxicity and glucose uptake assays in L6 myoblast cells, revealed that compounds 3d, 3g, and 3h significantly enhanced glucose uptake with low cytotoxicity22.
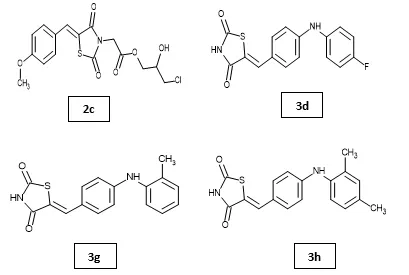
Shriram D. Ranade et al. developed acetyl pyrrolidine-2-carbonitrile based benzylidene thiazolidinedione hybrids (5a–5j) targeting PPAR-γ using integrated in silico, in vitro, and in vivo approaches. Compounds 5b and 5f, containing 4-hydroxybenzaldehyde and 4-fluorobenzaldehyde substitutions, respectively, showed significant glucose uptake activity comparable to pioglitazone along with dose-dependent PPAR-γ activation. Both compounds improved glucose homeostasis and reversed insulin resistance in fructose-induced rats, while compound 5b exhibited superior pharmacokinetic properties with better half-life and elimination profile23. Mohd. Javed Naim et al. synthesized and evaluated a series of 2,4-thiazolidinedione based amide derivatives for antidiabetic activity. Among the synthesized compounds, 6c, 6e, 6m, and 6n showed significant antidiabetic potential, with compound 6c bearing a 4-chlorophenyl substituent exhibiting the highest activity and a 2.1-fold increase in PPAR-γ gene expression compared to pioglitazone and rosiglitazone. Compound 6c also demonstrated favorable biochemical parameters without hepatotoxicity or significant weight gain, highlighting its potential as a safer antidiabetic agent24. Mohd. Javed Naim et al. designed and synthesized thiazolidinedione-based benzene sulphonamide derivatives containing a pyrazole core and evaluated their antidiabetic activity. Compounds 7b, 7d, and 7p showed significant antidiabetic potential, with compound 7p containing a naphthalene moiety exhibiting the highest activity and a 1.9-fold increase in PPAR-γ gene expression compared to pioglitazone and rosiglitazone25.
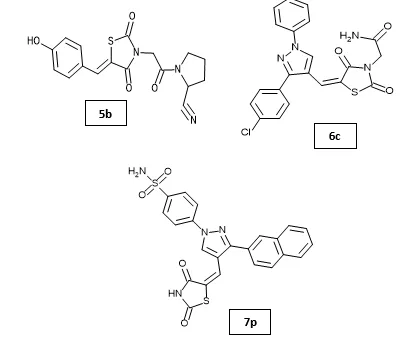
2.2 TZDs as anticancer agents
TZD derivatives have subsequently demonstrated promising anticancer effects against a wide range of cancer cell lines, including breast, liver, colorectal, lung, and prostate cancers26,27. The anticancer potential of TZDs is mediated through multiple mechanisms such as inhibition of cell proliferation, induction of apoptosis, cell cycle arrest, suppression of angiogenesis, and the induction of intrinsic apoptotic pathways6,28. In addition to PPAR-γ-dependent actions, several TZD derivatives exhibit anticancer activity through PPAR-γ-independent mechanisms involving mitochondrial dysfunction, caspase activation, VEGFR-2 inhibition, HDAC inhibition, and disruption of survival signaling pathways6,29-31. Furthermore, structural modification of the TZD scaffold and hybridization with other bioactive pharmacophores have significantly enhanced their cytotoxic potency, selectivity, and multitarget activity, highlighting TZDs as promising scaffolds for the development of novel anticancer agents28-31.
Pouria Zarrin et al. designed and synthesized novel thiazolidinedione (TZD) derivatives and evaluated their anticancer activity against MCF-7 breast cancer cells. Among the synthesized compounds, PZ-11 exhibited the most potent cytotoxic and antiproliferative activity, surpassing vincristine at lower concentrations. Gene expression studies revealed that PZ-11 induced apoptosis by downregulating antiapoptotic genes such as AIFM1, BAG3, and BIRC3 while upregulating pro-apoptotic markers including BAD, HRK, CASP10, and CASP14. Molecular docking and molecular dynamics simulations confirmed stable binding interactions of PZ-11 with apoptosis-inducing factor (AIF), supported by favorable ADMET properties. The study highlighted PZ-11 as a promising multitarget TZD-based anticancer lead with potential for breast cancer therapy through modulation of both intrinsic and extrinsic apoptotic pathways28. Khaled El-Adl et al. designed and synthesized a series of 5-benzylidenethiazolidine-2,4-dione derivatives targeting the VEGFR-2 enzyme and evaluated their anticancer activity against HepG2, HCT-116, and MCF-7 cancer cell lines. Among the synthesized compounds, 8f exhibited the most potent anticancer activity with IC50 values of 11.19, 8.99, and 7.10 μM against HepG2, HCT-116, and MCF-7 cells, respectively, showing activity comparable to sorafenib and doxorubicin against MCF-7 cells. Molecular docking studies revealed strong binding interactions with the VEGFR-2 active site, while VEGFR-2 inhibition assays demonstrated that compound 8f possessed the highest inhibitory activity (IC50 = 0.22 μM)29.
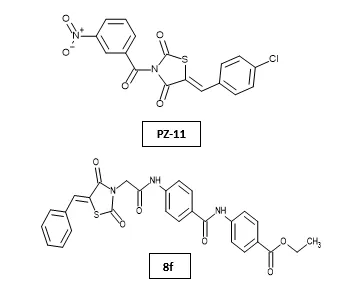
Mohammed S. Taghour et al. designed and synthesized a new series of thiazolidine-2,4-dione derivatives targeting VEGFR-2 as potential anticancer agents. The synthesized compounds were evaluated against A549, Caco-2, HepG-2, and MDA-MB-231 cancer cell lines, where compound 14a exhibited the most potent antiproliferative activity, particularly against Caco-2 and HepG-2 cells. VEGFR-2 kinase inhibition assays demonstrated significant inhibitory activity comparable to sorafenib. Further biological studies revealed that compound 14a effectively inhibited cancer cell migration and downregulated apoptosis-related markers, including Bcl-2, Survivin, and TGF. Molecular docking, MD simulation, and DFT studies confirmed stable binding interactions of 14a within the ATP-binding pocket of VEGFR-2, highlighting it as a promising lead compound for anticancer drug development30. Abdelgawad MA et al. designed and synthesized 12 novel thiazolidine-2,4-dione derivatives and evaluated their anticancer activity against HepG2, HCT-116, and MCF-7 cancer cell lines targeting VEGFR-2. Among the synthesized compounds, 7c and 6c exhibited the most potent activity against MCF-7 (IC50 = 7.78 and 8.15 µM), HCT116 (IC50 = 5.77 and 7.11 µM), and HepG2 (IC50 = 8.82 and 8.99 µM) cell lines, with better activity than sorafenib against HepG2 cells. These derivatives also showed strong VEGFR-2 inhibition (IC50 = 0.08 µM), significant PPAR-γ binding affinity (IC50 = 0.296 and 0.300 µM), and potent insulin-secreting activity (EC50 = 0.70 µM), along with low toxicity toward normal VERO cells. Molecular docking and ADMET studies further supported their dual role as VEGFR-2 inhibitors and PPAR-γ agonists6.
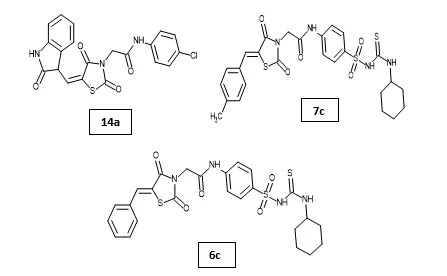
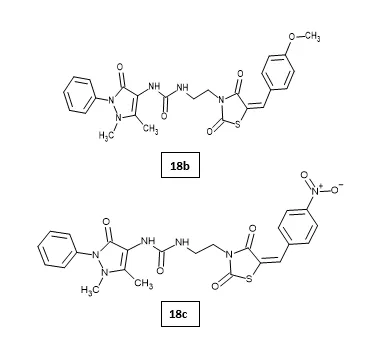
A. Hamdi et al. designed and synthesized novel 5-arylidenethiazolidine-2,4-dione hybrids and evaluated their antitumor activity against HePG2, HCT-116, MCF-7, PC3, and HeLa cancer cell lines. Compared to doxorubicin (IC50 = 4.17–8.87 μM) and SAHA (IC50 = 2.70–7.11 μM), compounds 18b, 18c, 18f, and 19d exhibited broad-spectrum antitumor activity against the selected cell lines with IC50 ranges of 3.16–24.14 μM. Compounds 18b and 18c showed potent HDAC inhibitory activity against HDAC1, HDAC2, HDAC6, and HDAC8 comparable to Entinostat. The apoptosis assay and cell cycle arrest analysis showed that derivative 18b induced apoptosis. Molecular docking analysis of the most active compounds 18b and 18c revealed good fitting and proper interactions with the major amino residues in the HDAC1, HDAC2, HDAC6, and HDAC8 binding sites. Based on these results, derivatives 18b and 18c are promising scaffolds for further modification and optimization to obtain potent antitumor agents with selective HDAC inhibitory activity31.
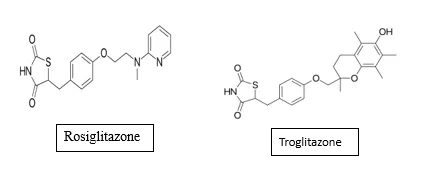
TZDs have attracted considerable interest for their anti-inflammatory effects beyond their antidiabetic activity. Activation of PPARγ suppresses the expression of pro-inflammatory mediators such as tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, inducible nitric oxide synthase (iNOS), and matrix metalloproteinases by modulating inflammatory signalling pathways including AP-1, NF-κB, STAT, and NFAT32-35. TZDs have demonstrated significant anti-inflammatory activity in various cell types and in animal models of inflammatory disorders such as rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, and asthma33,34. However, certain studies have also reported pro-inflammatory effects of TZDs such as Rosiglitazone and troglitazone in specific cell types, suggesting that their anti-inflammatory mechanisms may be complex and cell-type dependent35-37. These recent findings indicate a modulatory role for PPARs in inflammation with potential therapeutic applications in chronic inflammatory diseases32.
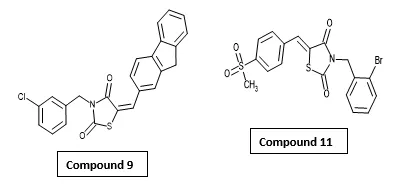
Cleiton Diniz Barros et al. synthesized a series of arylidene-thiazolidine-2,4-dione derivatives and evaluated their anti-inflammatory activity as potential Peroxisome Proliferator-Activated Receptor Gamma ligands. Among the synthesized compounds, compound 11 [3-(2-bromobenzyl)-5-(4-methanesulfonyl-benzylidene)-thiazolidine-2,4-dione] exhibited superior anti-inflammatory activity compared to the reference drug Rosiglitazone despite showing lower receptor affinity than rosiglitazone, suggesting that receptor affinity may not directly correlate with anti-inflammatory effects. Compound 9 also demonstrated significant in vivo anti-inflammatory activity despite not being confirmed as a direct PPARγ ligand. Furthermore, docking studies showed a good correlation with in vivo anti-inflammatory activity, supporting the potential of arylidene-thiazolidinediones as promising PPARγ-mediated anti-inflammatory agents35.
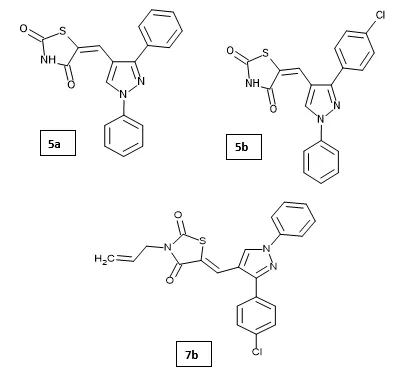
Amal M. Youssef et al. synthesized and evaluated a series of novel pyrazolyl-2,4-thiazolidinedione derivatives for their cytotoxic, anti-inflammatory, and neuroprotective activities. Among the synthesized compounds, derivatives 5a–c and 7b exhibited significant anti-inflammatory and neuroprotective potential with low cytotoxicity. In addition, compounds 5a–c inhibited the respiratory burst by targeting NADPH oxidase, thereby reducing the production of reactive oxygen species associated with inflammatory processes. Selected compounds (5a–c and 7b) further showed pronounced in vivo anti-inflammatory activity comparable to the reference drug Celecoxib in both acute and chronic inflammation models, along with excellent gastrointestinal safety and high tolerability in experimental animals. Among them, compound 5b showed superior activity in acute inflammation, whereas compound 5a was more effective against chronic inflammation36
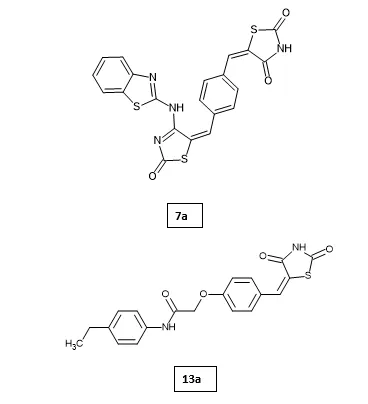
Shankar Gharge et al. designed and synthesized novel thiazolidine-2,4-dione derivatives via Knövenagel condensation and evaluated their antidiabetic, anti-inflammatory, and antioxidant activities. Among them, compound 13a exhibited significant blood glucose-lowering activity (108.5 ± 2.171 mg/dL), comparable to the standard drug Pioglitazone (101.66 ± 0.95 mg/dL). Additionally, compounds 7a and 13a demonstrated notable anti-inflammatory activity through COX-1 and COX-2 inhibition as well as strong antioxidant potential in DPPH and FRAP assays, surpassing standard ascorbic acid37.
CONCLUSION
Diabetes mellitus contributes to cancer development through chronic inflammation, insulin resistance, oxidative stress, and enhanced angiogenesis1,3,4. Current therapies often treat cancer and diabetes independently and are limited by toxicity, increased pill burden, poor patient compliance, limited target selectivity, and drug resistance2,4. Thiazolidinedione (TZD)-based derivatives have emerged as promising multifunctional scaffolds with significant therapeutic potential against diabetes, cancer, and inflammatory disorders6,7,8. Structural modification and molecular hybridization of the TZD nucleus with pharmacologically active moieties such as pyrazoles, quinazolinones, thiazoles, and sulfonamides have resulted in compounds with enhanced biological activity, target selectivity, and reduced toxicity. Numerous derivatives demonstrated potent antidiabetic activity through PPARγ modulation, anticancer effects via induction of apoptosis and inhibition of VEGFR-2 and HDAC enzymes, and anti-inflammatory properties through suppression of COX enzymes and inflammatory mediators22-34. Collectively, these findings highlight the versatility of TZD derivatives as valuable lead molecules for developing novel multifunctional therapeutic agents to treat diabetes-associated cancer and inflammation.
FUTURE PERSPECTIVES
Future research on thiazolidinedione (TZD)-based derivatives should focus on developing safer and more selective molecules with improved efficacy and reduced toxicity. Molecular hybridization with diverse bioactive scaffolds may enhance multitarget therapeutic potential against diabetes, cancer, and inflammatory disorders. Greater emphasis on structure–activity relationship studies, computational drug design, molecular docking, molecular dynamics simulations, and AI-assisted approaches may further optimize biological activity and target specificity. In addition, extensive in vivo and clinical studies, along with nanotechnology-based targeted drug delivery systems, may improve the bioavailability, safety, and therapeutic applicability of TZD-based agents.
REFERENCES


Neethu S.*, Seema Nair, Multitarget Therapeutic Potential of Thiazolidinedione-Based Derivatives in Cancer, Diabetes, and Inflammation: A Comprehensive Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5222-5235. https://doi.org/10.5281/zenodo.20772536
 10.5281/zenodo.20772536
10.5281/zenodo.20772536