
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacology, Channabasweshwar Pharmacy College, Latur
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia worldwide, neuropathologically defined by extracellular amyloid-? (A?) plaques, intracellular neurofibrillary tangles composed of hyperphosphorylated tau, and progressive synaptic and neuronal loss. Although amyloid and tau hypotheses have historically dominated AD research, increasing evidence positions chronic neuroinflammation as a central contributor to disease onset and progression rather than a secondary response. Activated microglia and reactive astrocytes sustain a pro-inflammatory milieu through release of cytokines, chemokines, reactive oxygen species, and complement proteins, thereby amplifying synaptic dysfunction and neuronal injury. A pivotal mechanism involves activation of the NLRP3 inflammasome, which promotes maturation of interleukin-1? and interleukin-18, linking innate immune activation to neurodegeneration. Therapeutic strategies are increasingly directed toward modulating glial phenotypes, inhibiting inflammasome signaling, and restoring immune homeostasis in parallel with amyloid- and tau-targeted approaches and disease modification strategies. However, translation of anti-inflammatory therapies has been limited by disease heterogeneity, suboptimal timing of intervention, and incomplete understanding of neuroimmune crosstalk. Contemporary integrative models suggest AD arises from interacting networks of proteostatic failure, immune dysregulation, and metabolic stress within the aging brain. This review summarizes current evidence on neuroinflammatory mechanisms in AD, evaluates emerging immunomodulatory therapies, and highlights future directions for precision-targeted interventions aimed at modifying disease progression, improving clinical trial design and therapeutic targeting, representing a major shift toward immune-centered therapeutic paradigms in Alzheimer’s disease research. Growing efforts also focus on identifying neuroinflammatory biomarkers in cerebrospinal fluid and blood to enable earlier diagnosis and patient stratification in clinical trials.
1.1 Alzheimer's Disease: A Global Health Challenge
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia worldwide, accounting for approximately 60-70% of all dementia cases [1]. Clinically, AD is characterized by a gradual decline in memory, cognition, language, executive function, and behavioral performance, ultimately resulting in loss of functional independence. The disease predominantly affects older adults, although rare early-onset familial forms have also been described. Despite substantial advances in understanding its molecular basis, AD remains one of the most challenging neurological disorders due to its complex and multifactorial pathogenesis [1].
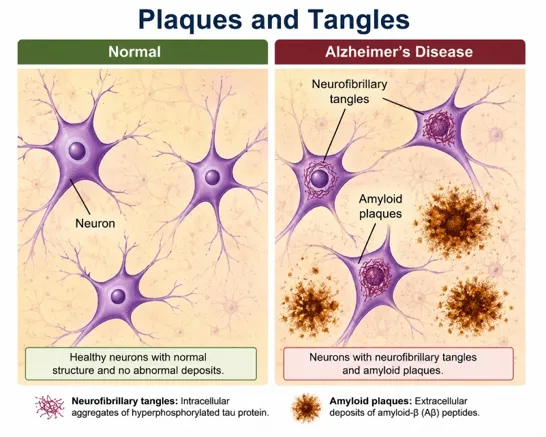
Figure 1. Structural brain changes in Alzheimer’s Disease
The global burden of AD has increased markedly over recent decades owing to demographic aging and increasing life expectancy. According to the World Health Organization, more than 55 million people are currently living with dementia worldwide, with projections indicating that this number may exceed 139 million by 2050 if effective preventive and therapeutic strategies are not implemented [2]. AD constitutes the largest proportion of these cases and has emerged as a major public health concern across both developed and developing nations. Epidemiological studies indicate that disease prevalence rises exponentially with advancing age, particularly after 65 years, making population aging a principal driver of future disease incidence [1,2]. Beyond its clinical implications, AD imposes a profound socioeconomic burden on patients, caregivers, healthcare systems, and society. The global cost associated with dementia care exceeds one trillion US dollars annually and continues to rise substantially with increasing disease prevalence [3]. In addition to direct healthcare expenditures, indirect costs related to long-term institutional care, productivity loss, and informal caregiving contribute significantly to the overall economic impact. Furthermore, caregivers frequently experience psychological distress, physical exhaustion, and reduced quality of life, highlighting the extensive societal consequences of the disease [4].
1.2 Classical Hallmarks of Alzheimer's Disease
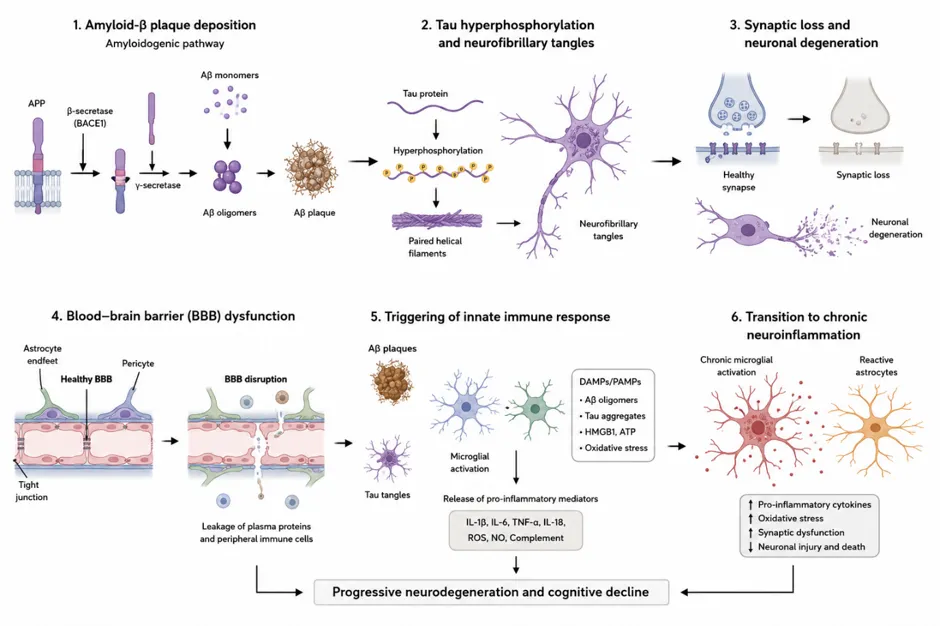
Figure 2: Pathological Hallmarks of Alzheimer’s Disease and Neuroinflammatory Cascade
Table 1. Classical Neuropathological Hallmarks of Alzheimer's Disease and Their Clinical Significance[5-8]
|
Hallmark |
Key Pathology |
Clinical Impact |
|
Amyloid plaques |
Extracellular Aβ42 aggregation |
Neuroinflammation and neuronal injury |
|
Neurofibrillary tangles |
Intracellular hyperphosphorylated tau |
Cognitive decline |
|
Synaptic dysfunction |
Aβ oligomer-mediated synaptic impairment |
Early memory deficits |
|
Neuronal loss |
Degeneration of hippocampal and cortical neurons |
Brain atrophy and dementia |
|
Braak staging |
Sequential spread of tau pathology |
Disease progression marker |
1.3 Emergence of Neuroinflammation as a Central Pathological Feature
Although AD has historically been viewed primarily as a disorder of protein aggregation, accumulating evidence suggests that neuroinflammation plays a fundamental role in disease initiation and progression [9,10]. Contemporary research indicates that inflammatory processes are not merely secondary responses to amyloid and tau pathology but active contributors to neurodegeneration. Microglia, the resident immune cells of the central nervous system, have emerged as critical regulators of AD pathogenesis. In the early stages of disease, microglia contribute to the clearance of amyloid deposits and cellular debris; however, chronic activation results in sustained production of pro-inflammatory cytokines, chemokines, reactive oxygen species, and other neurotoxic mediators that exacerbate neuronal injury [9,10]. The importance of innate immunity in AD is further supported by genome-wide association studies identifying several disease-associated risk genes, including triggering receptor expressed on myeloid cells 2 (TREM2), which is predominantly expressed in microglial populations [11].
Astrocytes also participate actively in neuroinflammatory responses through reactive astrogliosis, altered metabolic support, and dysregulated secretion of inflammatory mediators. Furthermore, growing evidence implicates inflammasome activation, particularly the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome, in amplifying neuroinflammatory signaling and promoting disease progression [12]. These findings have led to a paradigm shift in AD research, emphasizing complex interactions among amyloid pathology, tau aggregation, neuroimmune responses, and neuronal dysfunction.
1.4 Scope and Objectives of the Review
Given the growing recognition of neuroinflammation as a critical component of AD pathogenesis, a comprehensive understanding of the underlying inflammatory mechanisms has become increasingly important. This review aims to critically examine current knowledge regarding the role of neuroinflammation in AD, with particular emphasis on microglial and astrocytic activation, inflammatory signaling pathways, cytokine networks, inflammasome-mediated responses, and neuroimmune interactions. In addition, emerging therapeutic strategies targeting neuroinflammatory pathways and their translational potential will be discussed. By integrating evidence from experimental, translational, and clinical studies, this review seeks to provide a comprehensive perspective on the contribution of neuroinflammation to AD progression and its implications for future disease-modifying interventions.
2. Overview of Neuroinflammation in Alzheimer's Disease
2.1 Definition of Neuroinflammation
Neuroinflammation refers to the coordinated immune response within the central nervous system (CNS) mediated primarily by microglia, astrocytes, endothelial cells, perivascular macrophages, and infiltrating peripheral immune cells. Under physiological conditions, these cellular components maintain tissue homeostasis through pathogen surveillance, debris clearance, synaptic remodeling, and neurotrophic support. However, sustained activation of innate immune signaling pathways results in a chronic inflammatory milieu characterized by excessive production of cytokines, chemokines, reactive oxygen species (ROS), complement proteins, and other inflammatory mediators that contribute to neuronal dysfunction and degeneration [13,14,15].
2.2 Physiological versus Pathological Neuroinflammatory Responses
Physiological neuroinflammation is an adaptive and tightly regulated process essential for maintaining CNS integrity. Resting microglia continuously survey the neural environment and rapidly respond to tissue injury or infection. Following activation, microglia facilitate phagocytosis of cellular debris, secrete neuroprotective growth factors, and coordinate repair mechanisms that promote restoration of homeostasis [16]. Similarly, astrocytes contribute to blood-brain barrier (BBB) maintenance, metabolic support, and regulation of synaptic transmission. In contrast, pathological neuroinflammation is characterized by prolonged and dysregulated activation of glial cells. Chronic exposure to Aβ oligomers, fibrillar plaques, hyperphosphorylated tau aggregates, and damage-associated molecular patterns (DAMPs) promotes persistent activation of pattern-recognition receptors including Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), and triggering receptor expressed on myeloid cells 2 (TREM2)-associated signaling pathways [17,18]. Activated microglia subsequently release pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor-alpha (TNF-α), while astrocytes undergo reactive transformation and amplify inflammatory cascades. Although acute inflammatory responses may initially facilitate Aβ clearance, prolonged activation leads to impaired phagocytic capacity, oxidative stress, synaptic dysfunction, and neuronal injury. This transition from protective to detrimental inflammation represents a critical feature of AD pathogenesis and may explain why inflammatory responses become self-perpetuating during disease progression [19].
2.3 Evidence Supporting Neuroinflammation in Alzheimer's Disease
Table 2. Evidence Supporting Neuroinflammation in Alzheimer's Disease.[20-22]
|
Evidence Type |
Major Findings |
Significance |
|
Human & Genetic Studies |
Elevated inflammatory biomarkers (IL-1β, IL-6, TNF-α, YKL-40, sTREM2) and identification of immune-related risk genes (TREM2, CD33, CR1, ABI3, PLCG2, INPP5D). |
Supports a causal role of immune dysregulation in AD. |
|
Animal Studies |
APP/presenilin transgenic models exhibit microglial activation, increased cytokine production, and altered pathology following manipulation of TREM2 and NLRP3 pathways. |
Demonstrates mechanistic links between neuroinflammation and neurodegeneration. |
|
Imaging Studies |
TSPO-PET imaging reveals increased microglial activation in AD-vulnerable brain regions and correlates with disease progression. |
Provides in vivo evidence of ongoing neuroinflammation. |
|
Post-Mortem Studies |
Activated microglia, reactive astrocytes, complement proteins, inflammasome components, and disease-associated microglia (DAM) are enriched around AD lesions. |
Confirms direct association between neuroinflammation and AD pathology. |
2.4 Neuroinflammation as a Driver Rather Than a Consequence
Historically, neuroinflammation was considered a secondary reaction to amyloid deposition and neuronal death. However, accumulating evidence challenges this view. The identification of immune-related genetic risk factors suggests that dysregulated inflammatory signaling may precede overt pathology and influence disease initiation [23]. Furthermore, experimental studies demonstrate that inflammatory mediators can directly enhance amyloidogenic APP processing, promote tau phosphorylation, impair synaptic function, and disrupt BBB integrity, thereby accelerating neurodegeneration [27].
3. Cellular Mediators of Neuroinflammation in Alzheimer's Disease
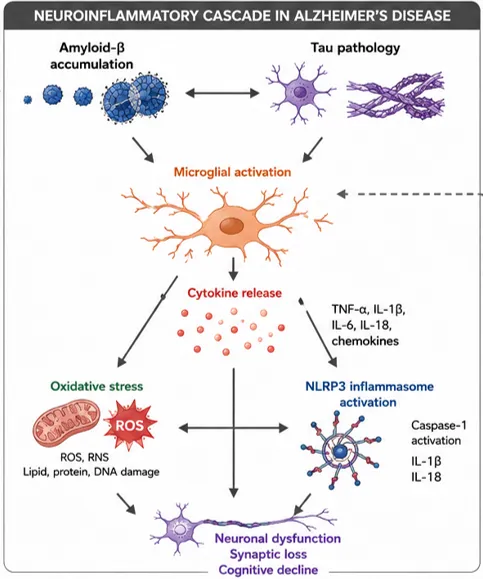
Fig 3. Neuroinflammatory cascade in Alzheimer’s disease
Neuroinflammation in Alzheimer's disease (AD) is mediated by a coordinated network of resident glial cells and infiltrating peripheral immune cells that collectively influence disease onset and progression. Among these cellular mediators, microglia and astrocytes represent the principal regulators of innate immune responses within the central nervous system (CNS), while oligodendrocytes and peripheral immune populations contribute to disease-associated inflammatory cascades. Although neuroinflammatory mechanisms may initially exert protective effects through the clearance of pathological proteins and maintenance of tissue homeostasis, chronic activation of these pathways promotes synaptic dysfunction, neuronal injury, and cognitive decline. Recent advances in transcriptomics, genetics, and neuroimaging have established neuroinflammation as a central pathogenic process rather than merely a secondary consequence of neurodegeneration [33].
Table 3. Cellular mediators of neuroinflammation in Alzheimer’s disease
|
Cell type |
Activation state in AD |
Key functions |
Contribution to pathology |
|
Microglia |
Homeostatic → DAM (Disease-Associated Microglia) |
Phagocytosis, immune surveillance, cytokine release |
Aβ plaque recognition, chronic cytokine secretion (IL-1β, TNF-α), synaptic pruning, NLRP3 inflammasome activation |
|
Astrocytes |
A1 (neurotoxic) / A2 (neuroprotective) |
BBB maintenance, neurotransmitter recycling, metabolic support |
A1 astrocytes release neurotoxic factors; impaired glutamate clearance; contribute to synaptic dysfunction |
|
Neurons |
Stress/degenerating neurons |
Synaptic transmission, signaling |
Release of DAMPs (damage-associated molecular patterns) triggering microglial activation; tau pathology propagation |
|
Oligodendrocytes |
Dysfunctional/ remyelination failure |
Myelin formation and axonal support |
White matter degeneration, impaired neuronal conduction, vulnerability to inflammation |
|
Endothelial cells (BBB unit) |
Activated/damaged |
BBB integrity, vascular transport regulation |
BBB breakdown → peripheral immune infiltration, plasma protein leakage, amplified neuroinflammation |
|
Peripheral macrophages/ monocytes |
Infiltrating immune cells |
Phagocytosis, cytokine production |
Enter CNS via compromised BBB; exacerbate inflammatory milieu |
3.1 Microglia
3.1.1 Microglial Physiology
Microglia are the resident immune cells of the CNS and originate from primitive yolk-sac macrophages during embryonic development. Under physiological conditions, they continuously survey the neural microenvironment through highly motile processes and participate in synaptic remodeling, neurogenesis, debris clearance, and maintenance of neuronal homeostasis [35,36]. Homeostatic microglia express characteristic markers such as P2RY12, TMEM119, and CX3CR1, which facilitate communication with neurons and other glial cells. Neuronal signaling molecules including fractalkine (CX3CL1) and CD200 contribute to the maintenance of a quiescent microglial phenotype and prevent excessive inflammatory activation [36].
3.1.2 Microglial Activation in AD
Microglial activation is among the earliest pathological alterations observed in AD. Amyloid-β (Aβ) oligomers and fibrils activate microglia through pattern-recognition receptors, including Toll-like receptors, scavenger receptors, and triggering receptor expressed on myeloid cells-2 (TREM2). Activation of these receptors initiates intracellular signaling pathways involving nuclear factor-kappa B (NF-κB), mitogen-activated protein kinases, and the NLRP3 inflammasome, resulting in the production of pro-inflammatory mediators [10,37].
Accumulating evidence indicates that microglia also respond to pathological tau species. Activated microglia contribute to tau propagation through the release of inflammatory mediators and extracellular vesicles, thereby linking innate immune activation with the spread of neurofibrillary pathology [38]. Furthermore, genome-wide association studies have identified several AD risk genes associated with microglial function, including TREM2, CD33, CR1, and PLCG2, highlighting the importance of innate immune dysregulation in disease susceptibility [15].
3.1.3 Protective Functions
Amyloid Clearance
Microglia play an important protective role during the early stages of AD by facilitating the recognition and removal of Aβ deposits. Through receptor-mediated mechanisms involving TREM2 and scavenger receptors, microglia internalize and degrade amyloid aggregates, thereby limiting plaque accumulation and associated neurotoxicity [18]. Experimental studies have demonstrated that functional TREM2 signaling promotes microglial migration toward amyloid plaques and enhances plaque compaction, reducing local neuronal damage [39].
Phagocytosis
In addition to amyloid clearance, microglia remove apoptotic cells, damaged synapses, and cellular debris through phagocytosis. This process is essential for maintaining CNS homeostasis and preventing the accumulation of potentially neurotoxic materials. However, aging and sustained inflammatory stimulation impair microglial phagocytic capacity, leading to reduced clearance efficiency and progressive accumulation of pathological substrates [10,18].
3.1.4 Detrimental Effects
Chronic Activation
Although acute microglial activation can be beneficial, persistent stimulation by Aβ and tau promotes a chronic inflammatory state characterized by metabolic dysfunction, oxidative stress, and exaggerated immune responses. Aging-associated microglial priming further enhances susceptibility to sustained activation, amplifying neuroinflammatory signaling and accelerating neuronal damage [9,40].
Cytokine Release
Chronically activated microglia release a broad range of pro-inflammatory cytokines and chemokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, and CCL2. These inflammatory mediators disrupt synaptic transmission, impair neuronal plasticity, alter blood-brain barrier integrity, and promote recruitment of additional immune cells into the CNS. Elevated levels of these cytokines have been consistently detected in AD brains and biological fluids, supporting their role in disease progression [9,17].
Synaptic Pruning
Recent studies have revealed that aberrant complement-mediated synaptic elimination represents a critical mechanism linking neuroinflammation to cognitive decline. Complement proteins C1q and C3 accumulate on vulnerable synapses, marking them for removal by microglia. Excessive synaptic pruning results in substantial synapse loss, which correlates strongly with cognitive impairment in AD [41,42].
3.1.5 Disease-Associated Microglia (DAM)
Single-cell transcriptomic studies have identified a specialized microglial phenotype termed disease-associated microglia (DAM). These cells exhibit reduced expression of homeostatic genes and increased expression of genes associated with lipid metabolism, phagocytosis, and inflammatory responses, including APOE, TREM2, TYROBP, and CST7 [43]. DAM appear to emerge in response to neurodegeneration and may initially contribute to amyloid containment and debris clearance. However, persistent DAM activation may promote chronic neuroinflammation and neurotoxicity, suggesting a dual role during disease progression [43,44].
3.2 Astrocytes
3.2.1 Normal Astrocytic Functions
Astrocytes are the most abundant glial cells in the CNS and perform numerous homeostatic functions. They regulate neurotransmitter recycling, maintain ion balance, support neuronal metabolism, preserve blood-brain barrier integrity, and participate in synaptic modulation. Astrocytes also secrete neurotrophic factors that promote neuronal survival and synaptic plasticity [45].
3.2.2 Reactive Astrogliosis
In AD, astrocytes undergo reactive astrogliosis, a process characterized by cellular hypertrophy, proliferation, and increased expression of glial fibrillary acidic protein (GFAP). Reactive astrocytes are frequently observed surrounding amyloid plaques and exhibit profound transcriptional and metabolic alterations [46]. Emerging evidence indicates that reactive astrocytes comprise heterogeneous populations with distinct functional properties. Some astrocytic phenotypes may retain protective functions, whereas others contribute directly to neurodegeneration through impaired metabolic support and enhanced inflammatory signaling [47].
3.2.3 Cytokine Production
Activated astrocytes produce a wide range of inflammatory mediators, including IL-1β, IL-6, TNF-α, transforming growth factor-β, and complement proteins. These molecules amplify local inflammatory responses and contribute to neuronal dysfunction. Notably, astrocytic production of complement component C3 has been associated with synaptic degeneration and cognitive decline in experimental models of AD [47,48].
3.2.4 Astrocyte-Microglia Crosstalk
Astrocytes and microglia engage in extensive bidirectional communication that regulates neuroinflammatory responses. Activated microglia release IL-1α, TNF-α, and C1q, which induce neurotoxic reactive astrocytes. In turn, astrocytes secrete cytokines and chemokines capable of further stimulating microglial activation. This reciprocal amplification loop contributes to chronic inflammation, synaptic dysfunction, and progressive neurodegeneration [47,49].
3.3 Oligodendrocytes and Myelin Dysfunction
Although less extensively studied than microglia and astrocytes, oligodendrocyte dysfunction has emerged as an important contributor to AD pathology. Oligodendrocytes are responsible for myelin formation and metabolic support of axons. Neuroimaging and neuropathological studies have demonstrated widespread white matter abnormalities and myelin degeneration in AD patients [50]. Inflammatory cytokines, oxidative stress, and amyloid-associated toxicity impair oligodendrocyte survival and remyelination capacity. Moreover, myelin debris can activate microglia and further exacerbate neuroinflammatory responses, establishing a self-perpetuating cycle of white matter damage and inflammation [50,51].
3.4 Peripheral Immune Cell Infiltration
Increasing evidence suggests that disruption of blood-brain barrier integrity during AD facilitates infiltration of peripheral immune cells into the CNS. These cells contribute to both protective and detrimental immune responses depending on disease stage and microenvironmental conditions [52].
Monocytes and Macrophages
Circulating monocytes are recruited into the CNS through chemokine-mediated pathways, particularly the CCL2-CCR2 axis. Following infiltration, these cells may differentiate into macrophages capable of phagocytosing Aβ deposits. However, prolonged recruitment can also enhance inflammatory signaling and tissue damage through cytokine production and antigen presentation [52].
T Lymphocytes
T lymphocytes have been identified in both the cerebrospinal fluid and brain parenchyma of AD patients. Recent studies have demonstrated expansion of clonally related cytotoxic CD8+ T-cell populations within the AD brain, suggesting active adaptive immune responses. These cells may influence disease progression through cytokine secretion and interactions with microglia and neurons [53].
Natural Killer Cells
Natural killer (NK) cells represent another peripheral immune population implicated in AD. Alterations in NK-cell phenotype and activity have been reported in affected individuals. Although their precise role remains incompletely understood, NK cells may modulate neuroinflammation through production of interferon-γ and interactions with resident immune cells [54]. Collectively, resident glial cells and infiltrating peripheral immune populations constitute a highly interconnected inflammatory network that influences virtually every stage of AD pathogenesis. Understanding the dynamic interplay among these cellular mediators is critical for the development of targeted immunomodulatory therapies aimed at limiting neuroinflammation while preserving essential homeostatic functions.
4. Molecular Mechanisms Underlying Neuroinflammation
4.1 Amyloid-β-Induced Inflammatory Signaling
Accumulation and aggregation of amyloid-β (Aβ) peptides represent one of the earliest pathological events associated with Alzheimer's disease (AD) and constitute a major trigger of innate immune activation within the central nervous system (CNS). Aβ is generated through sequential cleavage of amyloid precursor protein (APP) by β-secretase (BACE1) and γ-secretase, yielding peptides with varying lengths, among which Aβ42 exhibits the greatest propensity for oligomerization and fibrillization. Although soluble Aβ oligomers are increasingly recognized as the most neurotoxic species, insoluble fibrillar aggregates deposited as extracellular plaques also contribute significantly to chronic neuroinflammatory responses [55,56,]. Microglia recognize aggregated Aβ through multiple pattern-recognition receptors (PRRs), including toll-like receptors (TLRs), scavenger receptors, receptor for advanced glycation end products (RAGE), CD36, and triggering receptor expressed on myeloid cells 2 (TREM2). Binding of Aβ to these receptors initiates intracellular signaling cascades that activate transcription factors such as nuclear factor-kappa B (NF-κB), resulting in increased production of inflammatory mediators including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, IL-6, and reactive oxygen species (ROS) [9,33].
Initially, microglial activation promotes Aβ phagocytosis and clearance, constituting a protective response aimed at restoring tissue homeostasis [57]. However, persistent exposure to aggregated Aβ drives chronic activation of microglia, resulting in a shift toward a pro-inflammatory phenotype characterized by impaired phagocytic capacity and sustained cytokine production [9,18]. This maladaptive immune response establishes a self-perpetuating cycle in which neuroinflammation accelerates Aβ deposition, while increasing Aβ burden further amplifies inflammatory signaling [56]. Genetic studies identifying risk variants in immune-related genes such as TREM2, CD33, and CR1 further support the central role of immune dysfunction in Aβ-driven AD pathogenesis [15].
4.2 Tau-Mediated Neuroinflammation
While Aβ pathology is often considered the initiating event in AD, neurofibrillary pathology composed of hyperphosphorylated tau correlates more closely with neuronal dysfunction and cognitive decline. Tau is a microtubule-associated protein that normally stabilizes neuronal cytoskeletal architecture. Under pathological conditions, abnormal phosphorylation, truncation, and aggregation promote the formation of intracellular neurofibrillary tangles (NFTs) [58,59]. Emerging evidence indicates that pathological tau actively participates in neuroinflammatory processes rather than serving solely as a downstream consequence of disease progression. Extracellular tau released from damaged neurons can be recognized by microglial PRRs, including TLR2 and TLR4, stimulating inflammatory signaling pathways and cytokine secretion. Activated microglia subsequently release inflammatory mediators that enhance tau kinase activity, including glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent kinase 5 (CDK5), thereby promoting further tau hyperphosphorylation [59,60].
Tau pathology also exhibits prion-like propagation between anatomically connected brain regions [61]. Microglia appear to facilitate this process through uptake and release of tau-containing extracellular vesicles, contributing to the spread of pathology throughout neural networks [62]. Experimental studies demonstrate that suppression of microglial activation reduces tau propagation and mitigates neurodegeneration, highlighting a bidirectional relationship between tau pathology and innate immune activation [38].
4.3 Cytokine and Chemokine Networks
Table 4. Major Cytokines Involved in Neuroinflammation in Alzheimer's Disease[63-66]
|
Cytokine |
Primary Source |
Major Effects in AD |
|
TNF-α |
Microglia, astrocytes |
Promotes synaptic dysfunction, excitotoxicity, neuronal apoptosis, and Aβ production |
|
IL-1β |
Activated microglia |
Enhances neuroinflammation, microglial recruitment, astrocyte activation, and tau phosphorylation |
|
IL-6 |
Microglia, astrocytes |
Contributes to glial activation, impaired synaptic plasticity, and cognitive decline |
|
IL-10 |
Microglia, immune cells |
Suppresses excessive inflammation but may reduce Aβ clearance |
|
TGF-β |
Astrocytes, microglia |
Regulates immune homeostasis and tissue repair; dysregulation contributes to vascular and microglial dysfunction |
4.4 NLRP3 Inflammasome Activation
The NLRP3 inflammasome is a multiprotein complex consisting of NLRP3, ASC, and pro-caspase-1. Assembly of this complex results in activation of caspase-1, which cleaves pro-IL-1β and pro-IL-18 into their biologically active forms [67,28].
Activation of the NLRP3 inflammasome generally requires two signals. The priming signal is mediated through NF-κB activation following stimulation of TLRs or cytokine receptors, resulting in increased expression of NLRP3 and pro-inflammatory cytokines. A second activation signal is subsequently triggered by cellular stressors such as mitochondrial dysfunction, lysosomal rupture, potassium efflux, ROS generation, and aggregated Aβ [67,28].
Aβ fibrils promote NLRP3 activation following microglial phagocytosis and lysosomal destabilization. Activated inflammasomes increase IL-1β and IL-18 secretion, amplify neuroinflammation, and impair Aβ clearance mechanisms [67,68]. Studies in transgenic AD mouse models demonstrate that genetic deletion of NLRP3 or caspase-1 reduces amyloid burden, attenuates neuroinflammation, and improves cognitive performance [37].
Recent evidence further suggests that NLRP3 activation contributes to tau hyperphosphorylation and aggregation, thereby linking the two major pathological hallmarks of AD [,68]. Given its central role in neuroinflammatory amplification, NLRP3 has emerged as an attractive therapeutic target. Several experimental inhibitors have demonstrated efficacy in reducing neuroinflammation and improving cognitive outcomes in preclinical models [67,68].
4.6 Toll-Like Receptor Signaling
Toll-like receptors represent a major family of PRRs involved in recognition of pathogen-associated molecular patterns and damage-associated molecular patterns. In AD, endogenous danger signals such as Aβ and tau activate multiple TLR pathways [9]. TLR2 expression is increased in microglia surrounding amyloid plaques. Binding of aggregated Aβ to TLR2 initiates MyD88-dependent signaling, promoting production of inflammatory cytokines and chemokines. TLR4 similarly recognizes Aβ aggregates and contributes to activation of innate immune responses. Although TLR4-mediated signaling may facilitate Aβ clearance during early disease stages, chronic activation promotes neurotoxicity through excessive cytokine production and oxidative stress. Both TLR2 and TLR4 converge upon NF-κB signaling pathways, resulting in transcriptional upregulation of inflammatory genes including TNF-α, IL-1β, IL-6, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2) [9,33,31].
4.7 Oxidative Stress and Neuroinflammation
Oxidative stress is both a consequence and a driver of neuroinflammatory responses in AD. Activated microglia and astrocytes generate substantial amounts of ROS through NADPH oxidase activity, mitochondrial dysfunction, and inflammatory signaling pathways. Mitochondrial impairment is particularly important because neurons possess high energetic demands and limited antioxidant capacity. Aβ and tau pathology disrupt mitochondrial respiration, increase electron leakage, and enhance ROS generation. Excessive ROS damage proteins, lipids, and nucleic acids, leading to progressive cellular dysfunction and neuronal death.
Importantly, oxidative stress amplifies inflammatory signaling by activating NF-κB and NLRP3 inflammasome pathways. In turn, inflammatory cytokines further increase ROS production, establishing a vicious cycle that accelerates neurodegeneration[67,69].
4.8 Blood-Brain Barrier Dysfunction
The blood-brain barrier (BBB) is a specialized interface that maintains CNS homeostasis through selective regulation of molecular and cellular trafficking. Increasing evidence indicates that BBB dysfunction occurs early in AD and contributes directly to neuroinflammation. Aβ accumulation, oxidative stress, and chronic inflammation induce endothelial damage, leading to disruption of tight junction proteins such as claudin-5, occludin, and zonula occludens-1. Loss of BBB integrity permits leakage of plasma proteins and neurotoxic molecules into the brain parenchyma. Compromised BBB function also facilitates immune cell infiltration, including monocytes, macrophages, and lymphocytes. These infiltrating cells release cytokines, chemokines, and ROS that further exacerbate local inflammatory responses.
At a broader level, AD is increasingly viewed as a disorder of the neurovascular unit, which comprises endothelial cells, pericytes, astrocytes, microglia, neurons, and extracellular matrix components [70,71]. Dysfunction of this integrated system impairs cerebral blood flow, reduces Aβ clearance, and promotes chronic inflammation.
5. Genetic Regulators of Neuroinflammation in Alzheimer's Disease
Table 5. Neuroinflammation-related genetic risk factors in Alzheimer’s disease.[72-76]
|
Gene |
Protein/ function |
Effect on immune response |
Relevance to AD |
|
APOE ε4 |
Lipid transport, microglial regulation |
Impaired Aβ clearance, increased inflammation |
Strongest genetic risk factor for sporadic AD; enhances microglial activation |
|
TREM2 |
Microglial receptor |
Regulates phagocytosis and lipid sensing |
Loss-of-function → reduced Aβ clearance, increased plaque-associated inflammation |
|
CD33 |
Sialic acid-binding receptor |
Inhibits microglial phagocytosis |
Reduces Aβ uptake, promotes inflammatory phenotype |
|
CR1 |
Complement receptor |
Regulates complement cascade |
Excess complement activation → synaptic pruning and neurodegeneration |
|
CLU (ApoJ) |
Chaperone protein |
Modulates complement and lipid transport |
Impaired Aβ aggregation clearance; inflammation modulation |
|
PICALM |
Endocytosis regulator |
Affects microglial uptake mechanisms |
Impaired Aβ trafficking and clearance |
|
NLRP3 (indirect risk axis) |
Inflammasome sensor |
Activates IL-1β/IL-18 release |
Central mediator of chronic neuroinflammation |
Collectively, APOE4, TREM2, CD33, CR1, PLCG2, ABI3, and INPP5D converge on common biological pathways involving microglial activation, phagocytosis, complement regulation, and inflammatory signaling. The identification of these genes has fundamentally shifted the understanding of AD from a purely amyloid-centric disorder toward a complex neuroimmune disease in which dysregulated innate immunity plays a pivotal pathogenic role [15,72,76].
6. Neuroinflammation and Disease Progression
Neuroinflammation is now recognized as a central contributor to Alzheimer's disease (AD) progression, linking amyloid-β (Aβ) accumulation and tau pathology to synaptic dysfunction, neuronal loss, and cognitive decline. While innate immune responses may initially serve protective functions by facilitating the clearance of misfolded proteins, persistent activation of microglia and astrocytes promotes chronic inflammatory signaling that exacerbates neurodegeneration [9,34].
6.1 Synaptic Dysfunction
Synaptic impairment represents one of the earliest pathological events in AD and correlates more closely with cognitive decline than amyloid plaque burden alone. Activated microglia contribute to aberrant complement-mediated synaptic pruning, resulting in excessive elimination of functional synapses. Concurrently, inflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) disrupt synaptic plasticity, impair long-term potentiation, and alter neurotransmitter homeostasis. Chronic neuroinflammation further enhances the synaptotoxic effects of Aβ oligomers and pathological tau species, thereby accelerating neuronal network dysfunction [9,10].
6.2 Neuronal Death
Persistent inflammatory activation contributes substantially to neuronal injury and death in AD. Activated glial cells generate reactive oxygen species, nitric oxide, and pro-inflammatory mediators that induce oxidative stress, mitochondrial dysfunction, and cellular damage. In addition, activation of inflammatory pathways such as the NLRP3 inflammasome promotes the release of IL-1β and IL-18, amplifying local immune responses and facilitating neurodegenerative processes. Neuroinflammation also promotes tau hyperphosphorylation and aggregation, creating a pathological feedback loop in which neuronal injury further stimulates inflammatory activation [9,33].
6.3 Cognitive Impairment
The combined effects of synaptic dysfunction and neuronal loss ultimately manifest as progressive cognitive impairment. Neuroinflammatory mediators disrupt communication within hippocampal and cortical networks responsible for memory, learning, and executive function. Clinical and experimental studies have demonstrated associations between increased microglial activation, elevated inflammatory biomarkers, and accelerated cognitive decline. Consequently, neuroinflammation is increasingly viewed not merely as a secondary response to pathology but as an active participant in the progression of cognitive deficits in AD [9,10].
6.4 Neuroinflammation Across Disease Stages
Neuroinflammatory responses evolve throughout the course of AD. During preclinical AD, microglia may initially exert protective effects by clearing Aβ deposits and maintaining tissue homeostasis. However, sustained exposure to accumulating pathological proteins gradually shifts microglia toward a pro-inflammatory phenotype. In mild cognitive impairment (MCI), increasing activation of microglia and astrocytes is associated with early synaptic dysfunction, propagation of tau pathology, and the emergence of measurable cognitive deficits. Inflammatory alterations at this stage are thought to contribute significantly to disease transition and progression [10,34].
In advanced AD, chronic neuroinflammation becomes widespread and self-perpetuating. Extensive gliosis, persistent cytokine production, complement activation, and inflammasome signaling amplify neurodegeneration and cognitive deterioration. These processes establish a vicious cycle in which inflammation promotes protein aggregation and neuronal damage, while accumulating pathology further sustains inflammatory responses [9].
7. Therapeutic Opportunities Targeting Neuroinflammation
Table 6. Current and emerging anti-inflammatory therapies in Alzheimer’s disease[77-96]
|
Therapeutic class |
Example agents |
Mechanism of action |
Development status |
|
NSAIDs |
Ibuprofen, naproxen |
COX inhibition → reduced prostaglandins |
Clinical trials largely inconclusive in established AD |
|
Glucocorticoids |
Dexamethasone (limited use) |
Broad immunosuppression |
Not recommended long-term due to adverse CNS effects |
|
Cytokine inhibitors |
Anti-TNF agents (etanercept, adalimumab) |
Block TNF signaling |
Experimental; limited BBB penetration |
|
NLRP3 inflammasome inhibitors |
MCC950 (experimental) |
Blocks IL-1β/IL-18 activation |
Preclinical promise; translational studies ongoing |
|
Microglial modulators |
P2X7 antagonists, TREM2 agonists |
Shift microglia to homeostatic state |
Early-stage clinical/ preclinical |
|
Complement inhibitors |
C1q inhibitors, C5 blockers (eculizumab analogs) |
Reduce synaptic pruning and complement cascade |
Preclinical/ early translational |
|
IL-1 pathway inhibitors |
Anakinra |
IL-1 receptor blockade |
Repurposing under investigation |
|
Immunomodulatory biologics |
IVIG (intravenous immunoglobulin) |
Multi-target immune regulation |
Mixed clinical outcomes |
|
Natural anti-inflammatory compounds |
Curcumin, resveratrol |
NF-κB inhibition, antioxidant effects |
Preclinical/ adjunctive exploration |
|
Gene-targeted therapies |
TREM2 agonistic antibodies |
Restore microglial phagocytosis |
Emerging precision medicine approach |
8. Future Perspectives
Precision Medicine Approaches
The increasing recognition of Alzheimer's disease (AD) as a biologically heterogeneous disorder has prompted a shift toward precision medicine approaches that integrate genetic, molecular, imaging, and clinical information to guide individualized therapeutic interventions. Genetic studies have identified several AD-associated risk loci, including APOE, TREM2, and CD33, which influence amyloid metabolism, microglial activation, and neuroinflammatory responses [15,10]. These findings suggest that patients may exhibit distinct pathogenic trajectories and therefore differ in their response to therapeutic interventions. The integration of genomic profiling with fluid biomarkers and neuroimaging measures may facilitate patient stratification, improve treatment selection, and enhance the likelihood of therapeutic success.
Multi-Target Therapeutics
The limited efficacy of many single-target therapies highlights the need for treatment strategies capable of addressing the multifaceted nature of AD pathology. In addition to amyloid-β and tau accumulation, neuroinflammation, synaptic dysfunction, mitochondrial impairment, and vascular abnormalities contribute substantially to disease progression [9,56]. Consequently, multi-target therapeutic approaches that simultaneously modulate protein aggregation and inflammatory pathways are gaining increasing attention. Combination therapies and multi-target-directed agents may provide broader neuroprotective effects by targeting interconnected pathological mechanisms rather than individual molecular abnormalities.
Biomarker-Guided Treatment
Recent advances in biomarker research have transformed the diagnostic and therapeutic landscape of AD. Blood-based biomarkers, including phosphorylated tau isoforms, neurofilament light chain (NfL), and glial fibrillary acidic protein (GFAP), enable the detection of pathological changes during the preclinical and prodromal stages of disease [97]. Beyond diagnosis, these biomarkers have considerable potential for monitoring disease progression, evaluating treatment response, and guiding therapeutic decisions. Biomarker-informed approaches may also improve clinical trial design by facilitating the selection of biologically defined patient populations most likely to benefit from specific interventions [97,98].
Artificial Intelligence and Drug Discovery
Artificial intelligence (AI) and machine learning technologies are emerging as powerful tools for accelerating AD research and drug development. Computational approaches can integrate large-scale genomic, transcriptomic, proteomic, imaging, and clinical datasets to identify novel disease mechanisms and therapeutic targets [99]. AI-assisted drug discovery platforms have the potential to shorten development timelines, improve target validation, and facilitate drug repurposing efforts. Furthermore, predictive modeling may enhance clinical trial efficiency by identifying patient subgroups with distinct biological signatures and therapeutic responses.
Personalized Neuroimmunomodulation
Given the central role of neuroinflammation in AD pathogenesis, personalized neuro-immunomodulation represents a promising future therapeutic direction. Increasing evidence suggests that microglial activation states vary among individuals and across disease stages, resulting in heterogeneous inflammatory profiles [10,33]. Therapeutic strategies targeting microglial receptors, inflammasome signaling, complement pathways, or cytokine networks may therefore require individualized implementation based on biomarker-defined inflammatory phenotypes. Although considerable challenges remain in defining clinically relevant immune endotypes, advances in single-cell transcriptomics and biomarker discovery are expected to facilitate the development of tailored immunomodulatory interventions capable of restoring neuroimmune homeostasis while minimizing adverse effects.
CONCLUSION
Alzheimer's disease is a complex neurodegenerative disorder characterized by progressive cognitive decline and extensive neuronal dysfunction. Although amyloid-β plaques and neurofibrillary tau tangles remain defining pathological features, contemporary evidence indicates that neuroinflammation is a central contributor to disease initiation and progression rather than merely a secondary consequence of protein aggregation. Interactions among activated microglia, reactive astrocytes, complement components, inflammatory cytokines, and peripheral immune signals create a sustained inflammatory environment that promotes synaptic dysfunction, neuronal injury, and cognitive deterioration.
Recent advances in genetics and molecular neuroscience have substantially improved understanding of the mechanisms linking immune dysregulation to AD pathology. The identification of immune-associated susceptibility genes such as TREM2 and CD33 has strengthened the concept that altered innate immune responses are fundamental components of disease pathogenesis. Furthermore, growing evidence indicates that microglial dysfunction contributes not only to neuroinflammatory amplification but also to impaired clearance of pathological proteins and disruption of neuronal homeostasis.
Therapeutic development has consequently expanded beyond traditional amyloid-centered strategies toward interventions targeting inflammatory signaling pathways and neuroimmune mechanisms. Emerging approaches include modulation of microglial activation states, inhibition of inflammasome pathways, regulation of complement-mediated synaptic pruning, and restoration of beneficial immune functions. Simultaneously, advances in blood-based biomarkers and neuroimaging technologies have improved diagnostic accuracy and enabled earlier identification of disease-associated pathological processes.
Despite significant progress, important challenges remain. The biological heterogeneity of AD, incomplete understanding of disease-stage-specific inflammatory mechanisms, and limitations in translating preclinical findings into clinical efficacy continue to hinder therapeutic advancement. Future research should focus on integrating biomarker-guided patient stratification, precision medicine frameworks, artificial intelligence-driven drug discovery, and personalized neuroimmunomodulatory strategies. Such multidisciplinary approaches may facilitate the development of effective disease-modifying therapies and ultimately improve outcomes for individuals affected by Alzheimer's disease.
REFERENCES


Kiran Surwase, Dr. Padmaja Giram, Manke M. B., Shubham Turewale, Rushikesh Choudhari, Neuroinflammation in Alzheimer's Disease: Molecular Mechanisms and Therapeutic Opportunities, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 3157-3177. https://doi.org/10.5281/zenodo.21383431
 10.5281/zenodo.21383431
10.5281/zenodo.21383431