
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Dept. of Pharmacology, Appasaheb Birnale College of Pharmacy, Sangli – 416416, Maharashtra, India.
Chronic insomnia is increasingly understood as a disorder extending beyond behavioral and circadian dysregulation, with growing evidence highlighting a central role for neuroimmune mechanisms. This review examines the contribution of neuroinflammatory signaling to the pathophysiology of chronic insomnia, focusing on microglial activation and its impact on sleep-wake regulation. Under normal conditions, immune mediators such as cytokines participate in sleep homeostasis; however, persistent sleep disturbance promotes a shift toward sustained low-grade inflammation. Clinical and preclinical studies consistently report elevated pro-inflammatory markers, including interleukin-6 and tumor necrosis factor-?, alongside alterations in central immune activity, suggesting a bidirectional interaction between sleep disruption and inflammatory processes. Microglia serve as key mediators linking peripheral immune signals with central neural circuitry. Chronic activation of microglia engages intracellular pathways such as NF-?B, NLRP3 inflammasome, and MAPK signaling, leading to synaptic dysregulation, impaired sleep architecture, and associated cognitive and emotional disturbances. Additional factors, including blood-brain barrier dysfunction and systemic inflammation, further amplify this neuroimmune imbalance. While current pharmacological therapies primarily provide symptomatic relief, they do not adequately target these underlying mechanisms. Emerging approaches aimed at modulating neuroinflammatory pathways and restoring microglial homeostasis may offer more effective, mechanism-based strategies for the management of chronic insomnia.
Chronic insomnia is a prevalent and debilitating sleep disorder characterized by persistent difficulty initiating or maintaining sleep, early morning awakenings, or non-restorative sleep, occurring at least three nights per week for longer than three months despite adequate opportunity for sleep, and resulting in significant daytime distress or impairment1. Across global populations, estimates suggest that approximately 10-30% of adults experience chronic insomnia symptoms, with higher prevalence observed in older adults, women, and individuals with psychiatric or medical comorbidities2,3. The public health burden of insomnia extends beyond subjective sleep loss: it increases risk for metabolic syndrome, cardiovascular disease, mood disorders, cognitive impairment, and all-cause mortality, emphasizing the need for mechanistic understanding and novel interventions(4)(5). While classical models of insomnia emphasize behavioral, psychological, and circadian factors, there is growing recognition that sleep and immune systems are deeply interconnected. Sleep regulates, and is regulated by, components of the innate and adaptive immune responses(6). Chronic sleep loss elicits persistent low-grade inflammation, evident in elevated peripheral cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), which are implicated in disrupted sleep architecture and daytime dysfunction(7)(8). This interplay suggests a bidirectional relationship where neuroimmune activation is both a cause and consequence of sleep disturbance. Central to innate immune signaling in the brain are microglia, the resident immune cells of the central nervous system. Originating from yolk-sac progenitors and distributed throughout the brain, microglia continually surveil the neural microenvironment and respond to homeostatic perturbations by altering morphology, gene expression, and secretory profiles(9)(10). Microglia modulate synaptic pruning, neurotransmitter metabolism, and cytokine production processes that influence sleep regulation and neural network stability(11)(12). In the context of chronic sleep loss, microglial activation triggers pro-inflammatory pathways that impair neural circuitry involved in the sleep-wake cycle, including hypothalamic, thalamic, and brainstem networks. Despite mounting evidence linking neuroinflammatory signaling to sleep dysregulation, several critical gaps remain. First, most human studies have relied on peripheral inflammatory markers with limited insight into central nervous system immune dynamics(13). Second, the mechanistic contributions of specific microglial signaling cascades (e.g., NF-κB, NLRP3 inflammasome) in chronic insomnia remain under-explored. Third, translational pharmacological approaches targeting neuroimmune pathways are still in early stages. These gaps impede the development of targeted, mechanism-based therapies. Accordingly, this review aims to synthesize current evidence on neuroinflammatory signaling in chronic insomnia, with emphasis on microglial activation and downstream effects on sleep-wake regulation. We first outline the epidemiology and neurobiology of insomnia before examining how inflammatory pathways intersect with sleep physiology, ultimately highlighting potential therapeutic targets and future research directions.
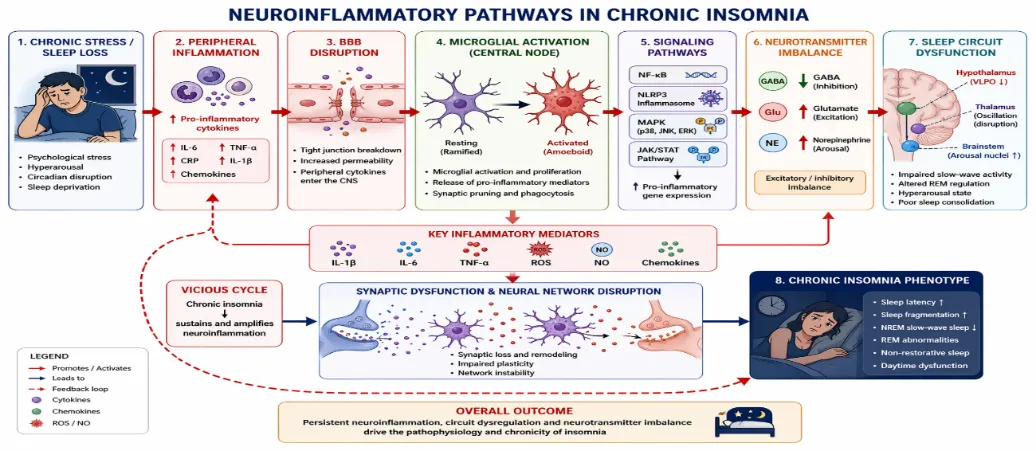
Figure 1 Neuroinflammatory Pathways in Chronic Insomnia
The Neuroimmune Interface In Sleep-Wake Regulation:
Understanding chronic insomnia requires first establishing the physiological architecture of sleep-wake control and its integration with immune signaling. Sleep is not merely a passive neurological state but a dynamic, actively regulated process governed by distributed neural circuits and modulated by immune mediators. Emerging evidence demonstrates that microglia and cytokine signaling are integral components of sleep homeostasis, thereby forming a neuroimmune interface that precedes pathological dysregulation.
Sleep Architecture And Regulatory Networks:
Neural Control of Sleep-Wake Cycles: Sleep-wake transitions are orchestrated through coordinated interactions among hypothalamic, brainstem, and thalamocortical networks. Classical models describe a “flip-flop” switch mechanism in which mutually inhibitory circuits promote stable wakefulness or sleep states(14). Wakefulness is sustained by monoaminergic and cholinergic neurons in the brainstem and hypothalamus, while sleep-promoting neurons in the ventrolateral preoptic area (VLPO) inhibit arousal systems to initiate non-rapid eye movement (NREM) sleep. Recent circuit-level analyses have refined this model, demonstrating that sleep state switching depends on dynamic integration of hypothalamic orexin neurons, basal forebrain cholinergic inputs, and ascending reticular activating pathways(15). Orexinergic projections stabilize wakefulness, whereas their attenuation facilitates sleep onset. Disruption in these stabilizing mechanisms increases state instability, a hallmark of insomnia.
Roles of the Hypothalamus, Brainstem, and Thalamus: The hypothalamus serves as a central integrator of circadian and homeostatic sleep signals. Sleep-promoting GABAergic neurons in the VLPO counterbalance wake-promoting histaminergic, serotonergic, and noradrenergic neurons(14). The brainstem reticular formation contributes to cortical activation and REM sleep regulation via cholinergic projections. The thalamus plays a critical role in shaping sleep architecture through thalamocortical oscillations. Slow waves and sleep spindles characteristic of NREM sleep arise from reciprocal thalamocortical interactions that regulate sensory gating and synaptic plasticity(16). These oscillatory mechanisms are sensitive to neuromodulatory tone, particularly norepinephrine, which declines during NREM sleep. Perturbations in thalamocortical coordination contribute to sleep fragmentation and reduced slow-wave activity observed in chronic insomnia.
Immune System Cross-Talk With The Central Nervous System:
Cytokines, Chemokines, and Glial Signaling: Sleep regulation is tightly coupled to immune mediators. Pro-inflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) exhibit somnogenic properties, enhancing NREM sleep when present at physiological levels(17,18). These cytokines fluctuate in a circadian pattern and interact with neuronal receptors to influence sleep intensity and duration. Chemokines and glial-derived signaling molecules further modulate synaptic activity and neuronal excitability. Astrocytes and microglia release cytokines and ATP in an activity-dependent manner, linking synaptic use to local immune signaling. This local network theory of sleep proposes that sleep emerges from activity-dependent inflammatory signaling within cortical columns(18).
Baseline Immune Signaling in Healthy Sleep: Physiological sleep is associated with tightly regulated, low-level immune activation. NREM sleep coincides with increased expression of certain cytokines that promote synaptic downscaling and restorative processes, whereas REM sleep involves suppression of pro-inflammatory tone(17). Importantly, this immune activation remains transient and homeostatically controlled. Disruption of sleep leads to amplification of inflammatory signaling beyond adaptive thresholds. Thus, the neuroimmune interface is not inherently pathological; rather, it is a regulatory mechanism that becomes maladaptive under chronic stress or sleep deprivation.
Microglia: Guardians Of The Cns
Microglial Origin, Phenotypes, and Activation States: Microglia originate from yolk sac progenitors during embryogenesis and persist as self-renewing resident immune cells of the central nervous system(9). In their surveillant state, microglia exhibit ramified morphology and continuously scan the neural environment. Their phenotypic plasticity allows rapid adaptation to environmental cues. Rather than conforming to a simplistic M1/M2 polarization model, microglia display a spectrum of activation states influenced by metabolic, neurotransmitter, and cytokine signals(19). Transcriptional profiling studies reveal diverse functional subsets associated with synaptic remodeling, immune surveillance, and inflammatory responses (11).
Homeostatic vs. Pro-Inflammatory Roles in Sleep: Under physiological conditions, microglia contribute to sleep regulation by modulating synaptic pruning and neurotransmitter balance. Recent evidence demonstrates that microglia regulate sleep architecture through calcium-dependent modulation of norepinephrine transmission, directly linking immune cells to arousal circuitry(12). This finding underscores that microglia are active participants in sleep-wake regulation rather than passive responders to injury.
However, sustained activation shifts microglia toward pro-inflammatory phenotypes characterized by elevated cytokine production, increased phagocytic activity, and altered morphology(10). Chronic activation disrupts synaptic homeostasis and perturbs hypothalamic and thalamocortical networks critical for stable sleep states. Thus, microglia represent a pivotal node in the neuroimmune interface balancing restorative signaling during healthy sleep while mediating pathological inflammation when regulatory mechanisms fail.
Chronic Insomnia And Neuroinflammatory Signaling:
Chronic insomnia is increasingly recognized as a disorder of sustained neuroimmune activation rather than solely a behavioral or circadian disturbance. Converging human and preclinical evidence demonstrates that persistent sleep disruption engages inflammatory signaling cascades within both peripheral and central compartments. These cascades converge on microglial activation, oxidative stress, blood-brain barrier (BBB) dysfunction, and intracellular pathways such as NF-κB and NLRP3, collectively destabilizing sleep-wake circuitry.
Evidence For Neuroinflammation In Insomnia
Human Clinical Studies: Clinical studies consistently demonstrate that individuals with chronic sleep disturbance exhibit elevated circulating inflammatory mediators, including IL-6, TNF-α, and C-reactive protein(7)(8)(20). Meta-analytic synthesis confirms that insomnia severity correlates with systemic low-grade inflammation, even after adjusting for confounders such as age, BMI, and depression(8). Neuroimaging and translational biomarker studies suggest that inflammatory signaling is not confined to the periphery. Sleep fragmentation and chronic insomnia phenotypes are associated with neuroimmune activation markers and altered central cytokine dynamics (21). These findings support a model in which peripheral inflammatory signals either cross or modulate the BBB to influence CNS microglial responses.
Animal Models of Sleep Disruption: Experimental sleep deprivation models provide mechanistic resolution. Rodent paradigms of chronic sleep restriction induce hippocampal and hypothalamic increases in IL-1β and TNF-α, accompanied by microglial hypertrophy and enhanced inflammatory gene expression(22)(21). Importantly, these inflammatory changes parallel impairments in memory consolidation and NREM slow-wave activity, suggesting causative linkage. Moreover, sleep loss impairs glymphatic clearance a sleep-dependent process critical for removal of metabolic waste thereby promoting accumulation of inflammatory mediators and reactive metabolites(23). Chronic disruption of this clearance system may amplify neuroinflammatory burden over time.
MICROGLIAL ACTIVATION IN CHRONIC INSOMNIA:
Microglia are central mediators of insomnia-associated neuroinflammation. Chronic sleep deprivation induces morphological transformation from ramified to amoeboid phenotypes, reflecting activation states associated with phagocytic and pro-inflammatory functions(22).
Activation Markers: Experimental models show upregulation of ionized calcium-binding adaptor molecule 1 (Iba1) and CD68 following prolonged sleep restriction, indicating enhanced microglial reactivity and lysosomal activity(22). These changes are region-specific, prominently affecting hippocampus, cortex, and hypothalamus regions integral to sleep regulation.
Functional Changes and Triggers: Sleep loss acts as a physiological stressor, increasing glucocorticoids and sympathetic tone, which in turn potentiate microglial inflammatory responses. Chronic activation alters synaptic pruning, neurotransmitter balance, and neuronal excitability, thereby destabilizing sleep-wake networks (21). Persistent microglial activation thus transforms adaptive immune surveillance into maladaptive neuroinflammation.
Key Pro-Inflammatory Mediators:
Cytokines: IL-1β, IL-6, TNF-α: IL-1β and TNF-α are somnogenic at physiological levels but become disruptive when chronically elevated. Sustained increases alter NREM architecture and increase sleep fragmentation(7)(20). IL-6 elevations correlate with insomnia severity and cardiometabolic risk, linking sleep disruption to systemic pathology(8).
Chemokines and Immune Amplification: Although less studied in insomnia specifically, chemokines coordinate leukocyte trafficking and microglial signaling, potentially reinforcing central inflammatory loops during prolonged sleep disturbance(21).
Reactive Oxygen and Nitrogen Species: Sleep deprivation induces oxidative stress characterized by increased reactive oxygen species (ROS) and nitric oxide derivatives in brain tissue2. Oxidative stress not only damages cellular components but also activates redox-sensitive transcription factors such as NF-κB, amplifying inflammatory gene expression.
Intracellular Signaling Pathways:
Chronic insomnia-associated neuroinflammation converges on several intracellular signaling cascades.
NF-κB Pathway: NF-κB is a master regulator of inflammatory transcription. Sleep deprivation activates NF-κB signaling in both peripheral leukocytes and CNS tissue, promoting expression of IL-1β, TNF-α, and adhesion molecules(19)(7). Persistent NF-κB activation sustains inflammatory tone and disrupts synaptic plasticity.
NLRP3 Inflammasome: The NLRP3 inflammasome is a cytosolic sensor complex that activates caspase-1 and promotes IL-1β maturation. Sleep restriction induces NLRP3 activation in hippocampal microglia, linking cellular stress to inflammatory amplification(24). NLRP3 is increasingly implicated in chronic neurodegenerative and inflammatory conditions, suggesting shared mechanistic pathways(25). Insomnia-driven metabolic stress and ROS production likely serve as upstream triggers.
MAPK Signaling (p38, JNK, ERK): Mitogen-activated protein kinases (MAPKs) regulate inflammatory gene expression and stress responses. Sleep disturbance activates p38 and JNK pathways in neural tissue, promoting cytokine transcription and apoptotic signaling(26). Dysregulated MAPK signaling may contribute to impaired synaptic homeostasis and altered REM/NREM balance.
TLRs and JAK/STAT Pathways: Toll-like receptor 4 (TLR4) signaling is upregulated following sleep deprivation, enhancing NF-κB activation and inflammatory cytokine release(27). Downstream, JAK/STAT signaling propagates cytokine receptor activation, further amplifying inflammatory responses in astrocytes and microglia(28). These cascades integrate peripheral immune cues with central inflammatory signaling.
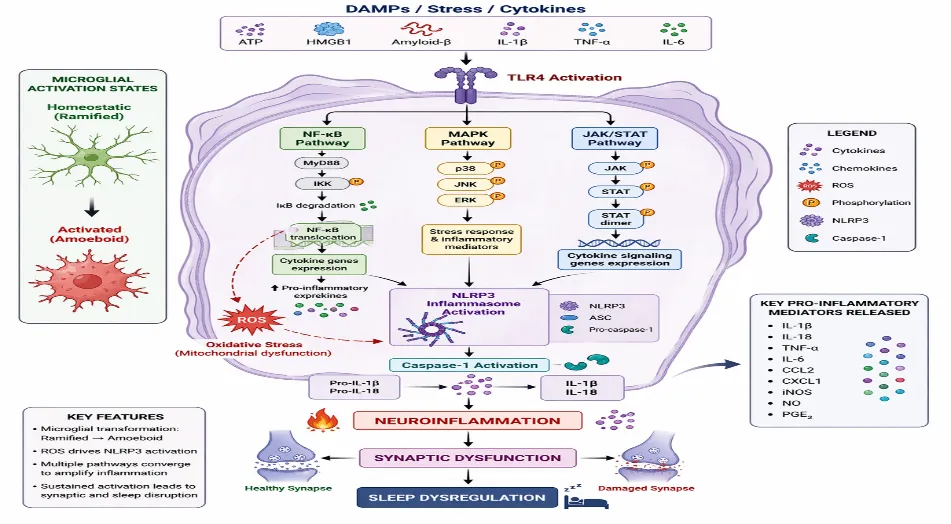
Figure 2 Microglial Activation & Signaling
Blood-Brain Barrier (Bbb) And Peripheral Influences:
BBB Dysfunction: Chronic sleep loss compromises BBB integrity, increasing permeability and facilitating peripheral cytokine entry into the CNS (29)(30). Tight junction protein disruption and endothelial inflammation represent key mechanisms.
Peripheral Immune Signals and CNS Impact: Elevated circulating cytokines may signal through endothelial receptors, vagal pathways, or direct paracellular transport, promoting microglial activation. Thus, insomnia-associated systemic inflammation directly influences central neuroimmune states(8).
Gut-Brain Axis: Although mechanistic evidence is emerging, sleep disruption alters gut microbiota composition, increasing endotoxin exposure and systemic inflammation, potentially engaging TLR4-dependent signaling in the brain(27). This axis may represent an underexplored amplifier of chronic insomnia-related neuroinflammation.
Sleep-Wake Dysregulation as a Functional Outcome
Neuroinflammatory activation in chronic insomnia is not a silent molecular phenomenon; it manifests as measurable alterations in sleep architecture, cognition, mood, and systemic physiology. The persistence of inflammatory signaling within sleep-wake regulatory circuits destabilizes neural oscillations, impairs synaptic plasticity, and disrupts homeostatic sleep processes. This section integrates mechanistic pathways discussed previously with functional outcomes at behavioral and systemic levels.
Alterations In Sleep Architecture:
Sleep Latency and Hyperarousal: Chronic insomnia is characterized by prolonged sleep latency and heightened physiological arousal. Neurobiological models describe insomnia as a disorder of cortical and subcortical hyperactivation, particularly within arousal-promoting circuits(31). Inflammatory cytokines such as IL-6 and TNF-α modulate hypothalamic and brainstem nuclei involved in sleep initiation, shifting the balance toward wake-promoting activity. Sleep deprivation studies demonstrate increased metabolic activity in wake-promoting networks and reduced inhibitory control from sleep-inducing regions(32). Persistent inflammatory signaling likely sustains this hyperarousal state via NF-κB and MAPK-dependent transcriptional programs that enhance neuronal excitability.
Sleep Fragmentation: Sleep fragmentation reflects instability in state switching mechanisms. Experimental sleep loss produces micro-arousals and reduced sleep efficiency, associated with inflammatory activation and altered thalamocortical oscillatory coherence(18). Cytokine imbalance disrupts slow-wave synchronization and spindle activity, undermining consolidated NREM sleep.
NREM/REM Alterations: Chronic insomnia is often associated with reduced slow-wave sleep (SWS) and altered REM density(31). Inflammatory mediators modulate NREM intensity under physiological conditions; however, chronic elevation produces dysregulated oscillatory activity. Neuroimaging of sleep deprivation reveals impaired glymphatic function and altered connectivity in limbic and prefrontal networks, further destabilizing REM-NREM cycling(32). These alterations reflect the downstream consequences of sustained neuroimmune activation within thalamocortical circuits.
Cognitive, Affective, And Behavioral Consequences:
Memory Impairment: Sleep plays a central role in memory consolidation, particularly hippocampal-dependent declarative memory and procedural learning. Sleep deprivation impairs long-term potentiation, synaptic plasticity, and hippocampal network stability(33). Neuroinflammation further exacerbates these impairments through cytokine-mediated suppression of neurogenesis and synaptic remodeling. Experimental evidence demonstrates that elevated inflammatory signaling correlates with deficits in attention, working memory, and executive function(32). Chronic insomnia therefore represents a state in which inflammation-driven synaptic dysregulation translates into measurable cognitive decline.
Mood Disturbances: Emotional regulation is highly sensitive to sleep loss. Reduced prefrontal-amygdala connectivity following sleep deprivation amplifies negative affect and stress reactivity(34). Pro-inflammatory cytokines modulate limbic circuits and monoaminergic systems, contributing to anxiety and depressive symptoms frequently comorbid with insomnia(31).
Inflammatory activation alters serotonergic and dopaminergic signaling, which are essential for mood stability. Thus, neuroinflammation provides a mechanistic bridge linking chronic insomnia to affective dysregulation.
Daytime Dysfunction: Daytime fatigue, reduced vigilance, and impaired occupational performance reflect cumulative neural inefficiency induced by sleep loss. Functional imaging studies reveal compensatory hyperactivation in frontoparietal networks following sleep restriction, indicative of reduced neural efficiency (32). Chronic inflammatory signaling likely compounds this inefficiency by disrupting mitochondrial function and promoting oxidative stress within cortical neurons.
Systemic Effects:
Metabolic Dysregulation: Chronic insomnia contributes to insulin resistance, glucose intolerance, and dysregulated appetite signaling. Sleep restriction alters leptin and ghrelin balance, increases sympathetic activation, and promotes systemic inflammation(35). Neuroinflammation interacts bidirectionally with metabolic pathways, as cytokines impair insulin signaling both centrally and peripherally. Persistent inflammatory activation thereby integrates sleep disturbance with obesity and metabolic syndrome risk.
Cardiovascular Risk: Sleep disruption is an independent risk factor for hypertension, coronary artery disease, and stroke(5). Elevated inflammatory cytokines and sympathetic overactivity promote endothelial dysfunction and vascular stiffness. Chronic insomnia-associated inflammation therefore extends beyond the CNS, contributing to systemic vascular pathology.
Immune Deviations: Sleep is essential for adaptive immune memory and immunological homeostasis. Experimental sleep deprivation reduces vaccine responsiveness and impairs immune memory consolidation(6). Chronic insomnia may therefore shift immune balance toward a pro-inflammatory yet functionally compromised state. Medic et al. (2017)(4) highlight that sustained sleep disruption amplifies susceptibility to infection while maintaining elevated inflammatory tone a paradoxical immune phenotype characterized by dysregulated host defense.
Integrative Perspective: Taken together, chronic insomnia-associated neuroinflammation destabilizes sleep architecture, impairs cognitive and emotional regulation, and drives systemic cardiometabolic risk. Inflammatory signaling disrupts thalamocortical oscillations, impairs hippocampal plasticity, and enhances limbic reactivity, translating molecular cascades into functional pathology. Thus, sleep-wake dysregulation is not merely a symptom of inflammation it is a direct physiological outcome of persistent neuroimmune activation.
Pharmacological Modulation Of Neuroinflammatory Pathways
Targeting neuroinflammatory signaling in chronic insomnia offers an opportunity to move beyond symptomatic sedation toward mechanism-based therapeutics. As evidence increasingly implicates microglial activation, inflammasome signaling, and cytokine dysregulation in sleep-wake instability, pharmacological strategies can be conceptualized across three tiers: 1 approved hypnotics with secondary immunomodulatory effects, 2 targeted intracellular pathway inhibitors, and 3 emerging immunomodulatory biologics and adjunctive agents. The translational challenge lies in restoring sleep homeostasis without disrupting physiological immune function.
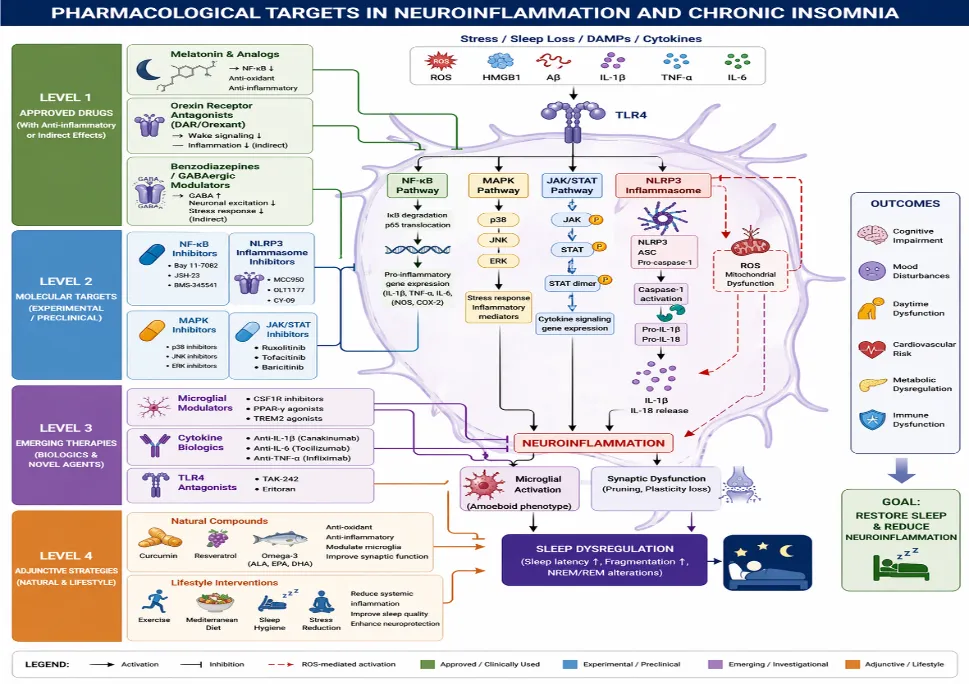
Figure 3 Pharmacological Targets
Approved Therapeutics With Anti-Inflammatory Properties:
Melatonin and Melatonin Receptor Agonists: Melatonin is not merely a chronobiotic hormone; it possesses significant anti-inflammatory and antioxidant properties. Mechanistically, melatonin suppresses NF-κB activation, reduces pro-inflammatory cytokine transcription, and inhibits NLRP3 inflammasome assembly.36,42 Through modulation of oxidative stress and mitochondrial function, melatonin attenuates ROS-mediated inflammatory amplification. Preclinical studies demonstrate that melatonin reduces microglial activation and improves sleep architecture under inflammatory conditions.36 These immunomodulatory effects may partially explain its benefit in insomnia phenotypes characterized by hyperarousal and inflammatory signaling.
Orexin Receptor Antagonists: Dual orexin receptor antagonists (DORAs) stabilize sleep-wake transitions by inhibiting wake-promoting orexin neurons. Clinical trials of daridorexant demonstrate improved sleep onset and maintenance with favorable next-day functioning .37 Lemborexant similarly shows sustained efficacy and acceptable safety in chronic insomnia.38 While primarily targeting arousal circuitry, orexin signaling intersects with stress and immune pathways. By reducing central hyperarousal and sympathetic tone, DORAs may indirectly attenuate inflammatory activation, although direct anti-inflammatory effects remain to be fully elucidated.39
Benzodiazepines and GABAergic Modulators: GABA-A receptor modulators reduce neuronal excitability and suppress hyperarousal. However, their immunomodulatory effects are indirect and largely limited to stress-axis attenuation. Long-term use is associated with tolerance and dependency risks, limiting their utility as anti-inflammatory strategies.39 Thus, while effective hypnotics, they do not directly target core inflammatory cascades implicated in chronic insomnia.
Targeting Intracellular Signaling Pathways:
Advances in neuroimmunology have identified specific intracellular cascades as potential pharmacological targets.
NF-κB Inhibitors: NF-κB is a master regulator of inflammatory gene transcription. Pharmacological inhibition reduces IL-1β, TNF-α, and IL-6 expression in preclinical models.40 Melatonin-mediated NF-κB suppression illustrates proof-of-concept for targeting this pathway in sleep-related inflammation.41 However, systemic NF-κB inhibition carries risks of impaired host defense.
NLRP3 Inflammasome Modulators: Selective NLRP3 inhibitors show promise in reducing microglial IL-1β maturation and neuroinflammatory burden.42 Targeting inflammasome activation may interrupt the feed-forward loop between oxidative stress and cytokine amplification central to chronic insomnia pathology. Because NLRP3 activation is stress- and ROS-dependent, its inhibition may be particularly relevant in sleep deprivation contexts.
MAPK Pathway Inhibitors: MAPKs (p38, JNK, ERK) regulate cytokine production and stress responses. Although not yet widely tested in insomnia, pharmacological targeting of these kinases has shown anti-inflammatory efficacy in neurodegenerative and stress-related models.40 Given MAPK activation during sleep loss, selective inhibition could theoretically stabilize inflammatory signaling without broad immunosuppression.
JAK/STAT Blockers: Cytokine receptor signaling often converges on JAK/STAT pathways. JAK inhibitors reduce inflammatory gene expression in CNS disorders and are under investigation for neuroimmune conditions.43 Modulating JAK/STAT signaling could attenuate cytokine-driven microglial activation in chronic insomnia, although CNS penetration and safety remain key concerns.
Emerging Small Molecules And Biologics:
Microglial Modulators: Targeting microglia directly represents a paradigm shift. Modulation of microglial phenotype rather than global suppression may restore homeostatic surveillance without impairing immune defense.44 Agents that reprogram microglia toward neuroprotective states could stabilize sleep-wake circuitry disrupted by inflammatory signaling.
Cytokine Blockers: Biologic agents targeting IL-1β or IL-6 pathways have demonstrated efficacy in systemic inflammatory diseases. While not yet established for insomnia, they provide mechanistic proof that cytokine blockade can normalize inflammatory cascades.40 Careful patient stratification would be required to justify such interventions in sleep disorders.
TLR Antagonists: Toll-like receptor (TLR) signaling contributes to sleep deprivation-induced inflammation. Targeting TLR4-mediated activation may reduce downstream NF-κB and inflammasome signaling. Although translational evidence remains preliminary, TLR antagonists represent a rational upstream intervention.
Adjunctive Strategies:
Natural Compounds: Curcumin suppresses NF-κB activation and reduces microglial cytokine production in experimental models.45 Resveratrol exerts antioxidant and anti-inflammatory effects via SIRT1 activation and NF-κB inhibition.46 Omega-3 fatty acids modulate inflammatory eicosanoid pathways and reduce cytokine production.47 Although bioavailability and dosing variability limit clinical translation, these compounds offer adjunctive strategies targeting oxidative and inflammatory drivers of insomnia.
Lifestyle Immunomodulation: Exercise and dietary interventions reduce systemic inflammation and improve sleep quality. Lifestyle modification may attenuate chronic inflammatory tone, indirectly stabilizing sleep architecture. These approaches provide low-risk adjuncts to pharmacotherapy.
Challenges And Safety Considerations:
CNS Penetration: Effective treatment requires agents capable of crossing the BBB and modulating central microglial activity. Many systemic anti-inflammatory drugs exhibit limited CNS penetration.
Off-Target Immunosuppression: Broad suppression of NF-κB, JAK/STAT, or cytokine pathways risks impairing host defense. Precision targeting of pathological, rather than physiological, inflammation is essential.
Balancing Sleep and Immune Function: Physiological sleep relies on controlled inflammatory signaling. Excessive suppression could disrupt adaptive immune processes and sleep homeostasis. Thus, therapeutic strategies must recalibrate not abolish neuroimmune activity.
Integrative Perspectives: Systems Biology And Biomarkers:
Understanding chronic insomnia through a purely reductionist lens isolating single cytokines or pathways is no longer sufficient. Neuroinflammation operates as a systems-level network integrating central neural circuits, peripheral immunity, metabolism, and behavior. Accordingly, multi-omics platforms and network-based biomarker models are emerging as critical tools to dissect the sleep-inflammation interface and to enable precision diagnostics.
MULTI-OMICS APPROACHES:
Transcriptomics: Transcriptomic profiling allows quantification of gene-expression changes across sleep-wake states and inflammatory conditions. Multi-omics frameworks integrating transcriptomics with proteomic and metabolomic layers reveal coordinated immune-metabolic reprogramming during sleep disruption.48 Sleep disturbance has been associated with altered expression of inflammatory genes, including NF-κB pathway components and cytokine transcripts, suggesting persistent immune activation signatures in chronic insomnia. Peripheral blood transcriptomic studies show upregulation of pro-inflammatory gene clusters following sleep restriction, supporting the concept that insomnia reflects a systemic inflammatory phenotype rather than a purely CNS-confined disorder.49
Proteomics: Proteomic approaches enable quantification of circulating cytokines, chemokines, complement factors, and neuroimmune mediators. Advances in high-throughput mass spectrometry have identified dysregulated inflammatory and synaptic proteins in individuals with sleep disorders.50 Proteomic signatures in insomnia frequently implicate IL-6-related pathways, acute-phase reactants, and oxidative stress mediators aligning with mechanistic findings from microglial activation studies.
Metabolomics: Metabolomic profiling provides insight into immune-metabolic coupling during sleep disruption. Altered lipid metabolites, kynurenine pathway intermediates, and mitochondrial stress markers have been documented in sleep-deprived states.51 These metabolic signatures may reflect microglial metabolic switching toward glycolysis during inflammatory activation, reinforcing the systems biology model where metabolic and immune signaling are inseparable in chronic insomnia. Collectively, multi-omics approaches enable integration of molecular layers rather than isolated biomarkers essential for identifying mechanistic clusters rather than single targets.
Biomarker Panels For Insomnia Diagnosis And Severity:
Single inflammatory markers lack specificity. A panel-based strategy is more realistic and clinically useful.
Peripheral Cytokines: Elevated circulating IL-6, TNF-α, and C-reactive protein have been associated with chronic insomnia severity and persistence.49 However, variability across cohorts limits their diagnostic reliability. Composite cytokine indices may improve sensitivity and reproducibility.
Microglial Activation Signatures: Positron emission tomography (PET) imaging targeting translocator protein (TSPO) enables in vivo assessment of microglial activation. Recent neuroimaging advances have refined the quantification of neuroinflammation in sleep-related conditions.52 Peripheral surrogates of microglial activation such as soluble TREM2 and exosome-derived inflammatory markers are under investigation as minimally invasive alternatives.13
Neuroimaging Biomarkers: Functional and molecular imaging studies reveal altered connectivity in thalamocortical and limbic circuits in chronic insomnia, potentially linked to inflammatory signaling.49 Integration of PET-based neuroinflammatory measures with fMRI-derived connectivity metrics may allow stratification of inflammatory insomnia phenotypes. The key shift here is moving from symptom-based diagnosis to biologically informed subtyping.
Network Models Of Sleep-Inflammation:
Sleep-inflammation interactions are bidirectional and nonlinear. Network modeling frameworks integrate neural circuitry, cytokine dynamics, metabolic flux, and behavioral outputs into computational systems.48
Systems biology models suggest:
Such models can identify central hubs e.g., NF-κB, NLRP3, orexin signaling whose perturbation disproportionately affects network stability. This approach may guide rational therapeutic targeting rather than trial-and-error pharmacology.
Gaps, Limitations, And Future Directions:
Despite growing mechanistic clarity linking chronic insomnia with neuroinflammatory signaling, the field remains fragmented, methodologically heterogeneous, and translationally immature. High-impact progress now depends less on identifying additional inflammatory molecules and more on resolving conceptual and methodological limitations that constrain reproducibility and clinical translation.
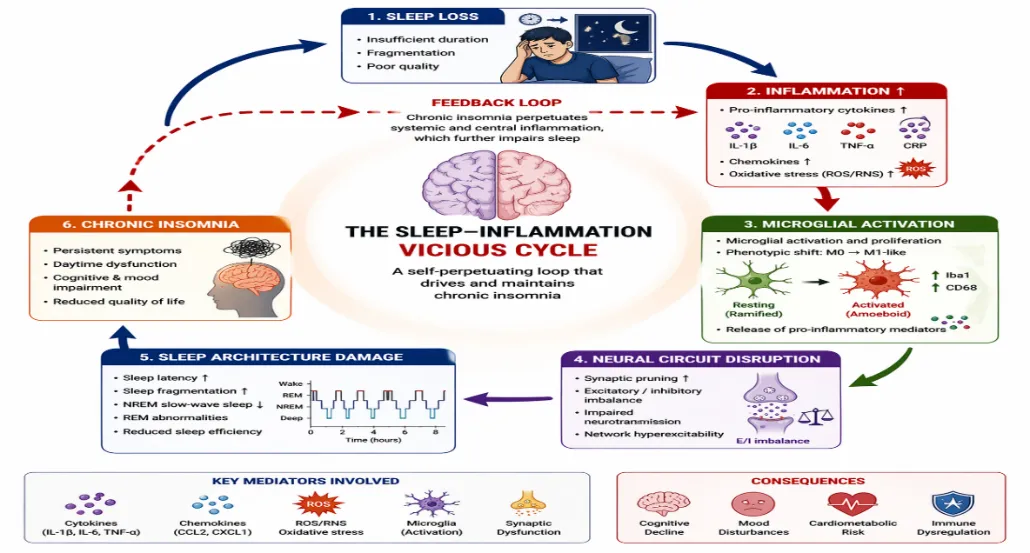
Figure 4 Sleep-Inflammation Feedback Loop
Need for Longitudinal Human Studies: Most human evidence linking insomnia and inflammation is cross-sectional. Elevated IL-6, TNF-α, or CRP levels are frequently reported, yet temporality remains unresolved: does inflammation drive insomnia, does insomnia amplify inflammation, or do both emerge from shared upstream stressors? Longitudinal cohort designs and intervention-based trials are required to determine causality and trajectory. Irwin et al. (2022)49 emphasize that repeated biomarker assessments across time combined with objective sleep metrics are necessary to identify inflammatory phenotypes predictive of chronicity, relapse, or treatment resistance. Without prospective data, mechanistic claims remain associative rather than causal. The field must shift from snapshot biology to dynamic immune profiling.
Standardized Inflammatory Readouts: Current studies use inconsistent sampling protocols, assay platforms, and biomarker panels, limiting comparability across cohorts. Cytokine concentrations are influenced by circadian phase, fasting state, stress exposure, and assay sensitivity. Feuth (2024)53 highlights the urgent need for harmonized protocols, including:
Without standardization, meta-analytic synthesis remains unreliable, and biomarker development for clinical use is unrealistic.
Species Differences in Microglial Responses: Rodent sleep deprivation models provide detailed mechanistic insights into microglial activation, inflammasome signaling, and synaptic remodeling. However, translational gaps persist. Picard et al. (2024)10 underscore that human microglia exhibit transcriptional and functional differences compared to murine models, including divergent cytokine profiles and receptor expression patterns. Similarly, inflammasome dynamics in neurodegenerative models may not directly map onto human insomnia pathology (Heneka et al., 2020)25. This species divergence raises a critical issue: mechanistic targets validated in rodents may not behave identically in humans. Advanced human-relevant systems induced pluripotent stem cell (iPSC)-derived microglia, organoids, and in vivo PET imaging must complement animal research to bridge translational gaps.
Precision Pharmacological Targets: Targeting “inflammation” as a broad construct is biologically naïve. Neuroimmune signaling is highly context-dependent, temporally regulated, and region-specific. Broad immunosuppression risks impairing physiological sleep-immune homeostasis.
Schwartz et al. (2021)44 argue for precision neuroimmunology approaches that:
Future pharmacology should focus on modulating maladaptive microglial activation states while preserving homeostatic surveillance functions. This demands biomarker-guided trials rather than symptom-based prescribing.
Clinical Trials Linking Inflammation Reduction to Sleep Restoration: A major translational weakness is the absence of robust randomized trials demonstrating that reducing neuroinflammation restores sleep architecture in chronic insomnia. Although anti-inflammatory agents improve inflammatory biomarkers, few studies simultaneously measure objective sleep outcomes (e.g., polysomnography, actigraphy) and central inflammatory markers. Irwin et al. (2022)49 highlight the need for mechanistic clinical trials incorporating:
Without demonstrating that immune modulation directly improves sleep physiology not merely mood or subjective distress the inflammatory hypothesis remains incomplete.
Future Research Priorities:
Mechanistic RCTs linking inflammation reduction with polysomnographic restoration.
REFERENCES

Saniya M. Mulla*, Shravani Patole, Shirish Patil, Dr. Padma. L. Ladda, Neuroinflammatory Signaling in Chronic Insomnia: From Microglial Activation to Sleep-Wake Dysregulation, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5095-5114. https://doi.org/10.5281/zenodo.20763061
 10.5281/zenodo.20763061
10.5281/zenodo.20763061