
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Matoshri College of Pharmacy, Eklahare, Nashik-422105.
Type 2 diabetes mellitus (T2DM) represents one of the most prevalent chronic metabolic disorders worldwide and continues to impose a substantial clinical and socioeconomic burden due to its progressive nature and associated complications. Among the emerging therapeutic options for T2DM management, sodium-glucose co-transporter-2 (SGLT2) inhibitors have gained significant attention owing to their insulin-independent mechanism of action and favorable effects on cardiovascular and renal outcomes. Canagliflozin, a first-generation SGLT2 inhibitor, effectively lowers plasma glucose concentrations by inhibiting renal glucose reabsorption in the proximal tubules, thereby enhancing urinary glucose excretion. However, the pharmaceutical development of canagliflozin is challenged by its poor aqueous solubility, low dissolution rate, and limited oral bioavailability, characteristics that contribute to variable absorption profiles and inconsistent therapeutic performance.The biopharmaceutical limitations of canagliflozin have stimulated considerable research interest in the development of advanced formulation strategies aimed at improving its solubility and dissolution characteristics. Among these approaches, solid dispersion technology has emerged as one of the most promising and extensively investigated techniques for enhancing the oral delivery of poorly water-soluble drugs. The incorporation of canagliflozin into hydrophilic carrier matrices facilitates the transformation of the drug from its crystalline state into a partially or completely amorphous form, resulting in improved wettability, reduced particle aggregation, enhanced surface area, and accelerated dissolution kinetics. Various hydrophilic polymers, including polyvinylpyrrolidone, polyethylene glycol, hydroxypropyl methylcellulose, and amphiphilic carriers, have demonstrated significant potential in improving the physicochemical properties and in vitro dissolution behavior of canagliflozin formulations.Furthermore, the incorporation of optimized solid dispersions into immediate-release tablet systems offers additional therapeutic advantages by promoting rapid tablet disintegration and prompt drug release following oral administration. Such formulations are particularly beneficial in antidiabetic therapy, where rapid attainment of effective plasma concentrations contributes to improved glycemic control and enhanced patient adherence. Nevertheless, successful formulation development requires a delicate balance between mechanical integrity, content uniformity, dissolution performance, and long-term stability. In this context, the application of statistical experimental designs, particularly factorial design methodologies, has become increasingly important in pharmaceutical product development. Factorial design enables systematic evaluation of formulation variables and their interaction effects on critical quality attributes such as drug release, disintegration time, tablet hardness, and friability. Moreover, integration of these statistical approaches within the Quality by Design (QbD) framework facilitates the identification of critical material attributes and process parameters, thereby ensuring robust product performance, regulatory compliance, and manufacturing reproducibility.This review critically summarizes the physicochemical characteristics and biopharmaceutical challenges associated with canagliflozin and comprehensively discusses contemporary formulation strategies with particular emphasis on solid dispersion technology, immediate-release dosage forms, and formulation optimization using factorial design principles. Additionally, recent advancements, emerging technologies, and future research opportunities aimed at improving the therapeutic efficiency and oral bioavailability of canagliflozin are highlighted. The integration of solubility enhancement techniques with scientifically optimized formulation approaches represents a promising pathway toward the development of effective and clinically reliable canagliflozin delivery systems for the management of type 2 diabetes mellitus.
1.1.OVERVIEW OF DIABETIC MELLITUS:
Diabetes mellitus (DM) is a chronic metabolic disorder characterized by persistent hyperglycemia resulting from defects in insulin secretion, insulin action, or a combination of both. The disease has emerged as one of the most significant public health challenges worldwide due to its increasing prevalence, associated comorbidities, and substantial economic burden on healthcare systems. Chronic elevation of blood glucose levels leads to progressive damage to various organs, particularly the kidneys, eyes, nerves, heart, and blood vessels, thereby contributing to increased morbidity and mortality among affected individuals. The pathophysiology of diabetes mellitus is complex and involves disturbances in carbohydrate, lipid, and protein metabolism. Under normal physiological conditions, insulin produced by pancreatic β-cells facilitates glucose uptake by peripheral tissues and maintains glucose homeostasis. In diabetes mellitus, impairment in insulin production or reduced cellular responsiveness to insulin results in elevated circulating glucose concentrations and subsequent metabolic dysfunction.
Diabetes mellitus is broadly classified into several categories, including Type 1 diabetes mellitus (T1DM), Type 2 diabetes mellitus (T2DM), gestational diabetes mellitus (GDM), and specific forms resulting from genetic defects, endocrine disorders, or drug-induced conditions. Type 1 diabetes mellitus is an autoimmune disorder characterized by destruction of pancreatic β-cells, leading to absolute insulin deficiency and lifelong dependence on exogenous insulin therapy. In contrast, Type 2 diabetes mellitus is characterized by a combination of insulin resistance and progressive β-cell dysfunction and accounts for approximately 90–95% of all diabetes cases globally.
Type 2 diabetes mellitus has become increasingly prevalent due to rapid urbanization, sedentary lifestyles, obesity, unhealthy dietary habits, and population aging. The disease is frequently associated with metabolic syndrome and is considered a major risk factor for cardiovascular diseases, chronic kidney disease, neuropathy, retinopathy, and peripheral vascular complications. Early diagnosis and effective glycemic control are therefore essential to minimize disease progression and reduce the incidence of long-term complications.
The management of T2DM involves lifestyle modifications, dietary interventions, regular physical activity, and pharmacological therapy aimed at achieving optimal glycemic control while minimizing adverse effects. Conventional antidiabetic agents such as metformin, sulfonylureas, thiazolidinediones, and dipeptidyl peptidase-4 inhibitors have been widely used; however, newer therapeutic classes have gained considerable importance due to their additional metabolic and organ-protective benefits.
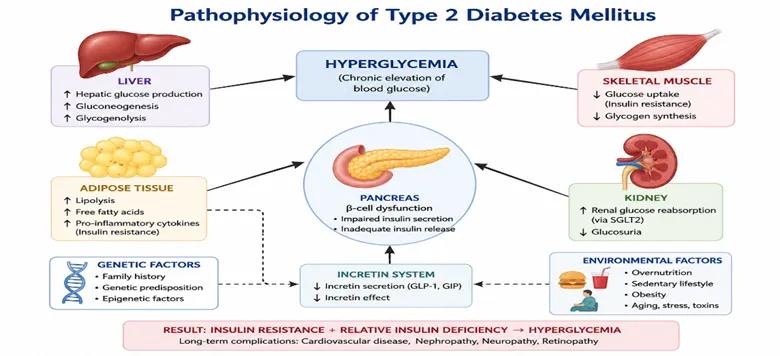
FIG. 1. Pathophysiology of type 2 diabetes mellitus
Among these, sodium-glucose co-transporter-2 (SGLT2) inhibitors have emerged as an important class of oral antihyperglycemic agents that lower blood glucose concentrations through an insulin-independent mechanism. These agents inhibit glucose reabsorption in the proximal renal tubules and promote urinary glucose excretion, thereby improving glycemic control while simultaneously providing cardiovascular and renal protective effects. Canagliflozin, one of the first clinically approved SGLT2 inhibitors, has demonstrated significant efficacy in the management of T2DM and has become an important therapeutic option for patients requiring comprehensive metabolic control. Despite its clinical advantages, the pharmaceutical development of canagliflozin remains challenging because of its poor aqueous solubility and limited oral bioavailability, necessitating the development of advanced formulation strategies to improve its therapeutic performance. This has led to growing interest in solubility enhancement technologies such as solid dispersion systems and optimized immediate-release formulations for improving the oral delivery of canagliflozin in the treatment of Type 2 diabetes mellitus.
1.2. burden of type 2 diabetes mellitus:
Type 2 diabetes mellitus (T2DM) has emerged as one of the most significant non-communicable diseases worldwide and represents a major public health challenge due to its rapidly increasing prevalence, chronic nature, and associated complications. The disease is characterized by insulin resistance and progressive pancreatic β-cell dysfunction, resulting in persistent hyperglycemia and disturbances in carbohydrate, lipid, and protein metabolism. The growing burden of T2DM is primarily attributed to rapid urbanization, sedentary lifestyles, unhealthy dietary habits, increasing obesity rates, population aging, and genetic susceptibility. T2DM accounts for approximately 90–95% of all diabetes cases and has shown a continuous rise in both developed and developing countries. The increasing prevalence of obesity and metabolic syndrome has further accelerated disease incidence, particularly among younger populations who were previously considered at low risk. This epidemiological transition has transformed T2DM from a disease predominantly affecting older adults into a major health concern across all age groups.
The clinical burden of T2DM extends beyond hyperglycemia and includes a wide spectrum of microvascular and macrovascular complications. Chronic uncontrolled blood glucose levels contribute to the development of diabetic nephropathy, retinopathy, neuropathy, and peripheral vascular disease, which significantly impair quality of life and increase healthcare utilization. Furthermore, patients with T2DM are at substantially higher risk of cardiovascular diseases, including coronary artery disease, myocardial infarction, heart failure, and stroke, which remain the leading causes of mortality among diabetic individuals.
In addition to its clinical consequences, T2DM imposes a considerable economic burden on healthcare systems due to long-term pharmacotherapy, frequent hospitalizations, management of complications, and loss of productivity. The increasing demand for effective and affordable therapeutic interventions has therefore become a priority for healthcare providers, researchers, and regulatory agencies worldwide.
Among the contemporary therapeutic approaches, sodium-glucose co-transporter-2 (SGLT2) inhibitors have gained considerable importance because of their ability to improve glycemic control through an insulin-independent mechanism while simultaneously providing cardiovascular and renal protective benefits. Canagliflozin, a selective SGLT2 inhibitor, lowers blood glucose concentrations by inhibiting renal glucose reabsorption in the proximal tubules and promoting urinary glucose excretion. However, despite its therapeutic efficacy, canagliflozin exhibits poor aqueous solubility and limited oral bioavailability due to its BCS Class IV characteristics, which present significant formulation challenges and justify the need for advanced drug delivery approaches such as solid dispersion systems and optimized immediate-release formulations.
1.3. Role of SGLT2 Inhibitors:
Sodium-glucose co-transporter-2 (SGLT2) inhibitors represent a novel class of oral antihyperglycemic agents that have significantly transformed the therapeutic management of Type 2 Diabetes Mellitus (T2DM). Unlike conventional antidiabetic medications that primarily act by stimulating insulin secretion or improving insulin sensitivity, SGLT2 inhibitors reduce plasma glucose concentrations through an insulin-independent mechanism by targeting renal glucose handling. This unique mode of action provides effective glycemic control while minimizing the risk of hypoglycemia and preserving pancreatic β-cell function. Under normal physiological conditions, approximately 180 g of glucose is filtered daily by the renal glomeruli, of which nearly 90% is reabsorbed in the proximal convoluted tubules through the action of sodium-glucose co-transporter-2 (SGLT2) proteins. These transporters play a critical role in maintaining glucose homeostasis by preventing the loss of glucose through urine. In patients with T2DM, increased renal glucose reabsorption contributes to persistent hyperglycemia and exacerbates disease progression. SGLT2 inhibitors selectively block these transporters, thereby reducing the renal threshold for glucose reabsorption and promoting urinary glucose excretion, ultimately leading to a reduction in circulating blood glucose levels. This mechanism operates independently of insulin secretion or insulin sensitivity, making SGLT2 inhibitors effective across various stages of T2DM progression.
In addition to improving glycemic control, SGLT2 inhibitors provide several pleiotropic therapeutic benefits that extend beyond glucose lowering. Clinical studies have demonstrated favorable effects on body weight reduction, blood pressure control, and cardiovascular risk reduction. Furthermore, these agents exhibit renoprotective properties by reducing albuminuria, slowing the progression of diabetic nephropathy, and preserving renal function in patients with chronic kidney disease. Such multidimensional benefits have established SGLT2 inhibitors as an integral component of modern diabetes management guidelines.
Among the currently available SGLT2 inhibitors, canagliflozin has emerged as one of the first clinically approved agents and has demonstrated significant efficacy in improving glycaemic control in patients with T2DM. Canagliflozin exerts its pharmacological action by selectively inhibiting SGLT2 transporters located in the proximal renal tubules, thereby increasing urinary glucose excretion and lowering plasma glucose concentrations independently of insulin action. Additionally, canagliflozin has been associated with beneficial cardiovascular and renal outcomes, further enhancing its clinical utility in diabetic patients with comorbid conditions.
Despite its therapeutic advantages, the pharmaceutical development of canagliflozin is challenged by its poor aqueous solubility, low dissolution rate, and limited oral bioavailability due to its Biopharmaceutics Classification System (BCS) Class IV characteristics. These physicochemical limitations may lead to variability in drug absorption and therapeutic response following oral administration. Consequently, considerable research efforts have focused on the development of advanced formulation strategies, including solid dispersion systems and immediate-release tablet formulations, to enhance the dissolution behavior and bioavailability of canagliflozin and thereby optimize its clinical performance.
1.4. Introduction to Canagliflozin:
Canagliflozin is an orally administered antihyperglycemic agent belonging to the class of sodium-glucose co-transporter-2 (SGLT2) inhibitors, which are widely used in the management of Type 2 Diabetes Mellitus (T2DM). It was among the first SGLT2 inhibitors approved for clinical use and has gained considerable attention due to its unique insulin-independent mechanism of action and its additional cardiovascular and renal protective effects. By selectively inhibiting SGLT2 transporters located in the proximal renal tubules, canagliflozin reduces renal glucose reabsorption and enhances urinary glucose excretion, thereby lowering plasma glucose concentrations and improving glycemic control. This therapeutic approach differs fundamentally from conventional antidiabetic agents that primarily rely on stimulating insulin secretion or enhancing insulin sensitivity.
Chemically, canagliflozin is designated as (2S,3R,4R,5S,6R)-2-[3-[[5-(4-fluorophenyl)thiophen-2-yl]methyl]-4-methylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol and possesses a molecular weight of 453.52 g/mol. The drug is typically obtained as a white to off-white crystalline powder with a melting point ranging from approximately 95°C to 105°C. Canagliflozin exhibits poor aqueous solubility but demonstrates high solubility in organic solvents such as methanol and dimethyl sulfoxide. These physicochemical characteristics significantly influence its dissolution behavior and oral absorption profile.
According to the Biopharmaceutics Classification System (BCS), canagliflozin is classified as a Class IV drug, characterized by both low aqueous solubility and low permeability. Furthermore, the drug exists predominantly in a crystalline form, which contributes to its slow dissolution rate and limited oral bioavailability. Following oral administration, canagliflozin exhibits an absolute bioavailability of less than 65%, partly due to poor solubility and hepatic first-pass metabolism. These biopharmaceutical limitations represent significant challenges in the development of conventional oral dosage forms and may lead to variability in drug absorption and therapeutic efficacy. Pharmacologically, canagliflozin lowers blood glucose levels by inhibiting approximately 90% of glucose reabsorption mediated by SGLT2 transporters in the kidneys. By reducing the renal threshold for glucose reabsorption, the drug facilitates glucose elimination through urine and thereby improves glycemic control independently of insulin action. In addition to its glucose-lowering effect, canagliflozin has demonstrated beneficial effects on body weight reduction, blood pressure control, and the prevention of cardiovascular and renal complications associated with diabetes mellitus. Consequently, the drug has become an important therapeutic option for patients with T2DM, particularly those with coexisting cardiovascular or renal disease.
Despite its significant therapeutic advantages, the pharmaceutical development of canagliflozin remains challenging due to its unfavorable physicochemical properties. The poor aqueous solubility, low permeability, and extensive crystallinity of the drug necessitate the development of advanced formulation strategies aimed at improving dissolution characteristics and oral bioavailability. Among these approaches, solid dispersion technology has emerged as a promising technique capable of converting the crystalline drug into an amorphous form, thereby enhancing wettability, dissolution rate, and systemic exposure. The incorporation of such optimized solid dispersions into immediate-release tablet formulations offers an effective strategy for improving the therapeutic performance of canagliflozin in the management of Type 2 Diabetes Mellitus.
1.5. Need for Improved Formulations of Canagliflozin
Despite the proven clinical efficacy of canagliflozin in the management of Type 2 Diabetes Mellitus (T2DM), its therapeutic performance following oral administration is significantly influenced by several unfavorable physicochemical and biopharmaceutical properties. The major limitation associated with canagliflozin is its poor aqueous solubility, which results in a slow dissolution rate in gastrointestinal fluids and consequently limits the amount of drug available for absorption. Since dissolution is often the rate-limiting step for orally administered poorly water-soluble drugs, inadequate dissolution can lead to variability in plasma drug concentrations and inconsistent therapeutic outcomes.
According to the Biopharmaceutics Classification System (BCS), canagliflozin is categorized as a Class IV drug, exhibiting both low solubility and low permeability. This classification presents significant challenges for the development of conventional oral dosage forms because both dissolution and membrane permeation contribute to limited bioavailability. Furthermore, the crystalline nature of canagliflozin further reduces its dissolution efficiency by restricting molecular mobility and drug release in aqueous environments. Consequently, the oral absorption of the drug may become highly dependent on gastrointestinal conditions, including pH, gastric emptying time, intestinal motility, and food intake, resulting in interpatient variability in therapeutic response. In addition to poor solubility, canagliflozin undergoes hepatic first-pass metabolism, which further decreases the amount of active drug reaching systemic circulation. The absolute oral bioavailability of crystalline canagliflozin has been reported to be less than 65%, emphasizing the necessity for formulation approaches capable of improving dissolution characteristics and enhancing systemic exposure following oral administration.
To overcome these limitations, considerable research efforts have focused on the development of advanced drug delivery systems and formulation strategies aimed at improving the solubility, dissolution rate, and bioavailability of canagliflozin. Various techniques including particle size reduction, complexation, lipid-based delivery systems, nanotechnology-based formulations, and solid dispersion systems have been investigated for their potential to enhance oral absorption. Among these approaches, solid dispersion technology has emerged as one of the most promising strategies due to its ability to convert the drug from a crystalline state into an amorphous or molecularly dispersed form, thereby improving wettability, increasing surface area, and accelerating dissolution kinetics.
Furthermore, the incorporation of optimized solid dispersions into immediate-release tablet formulations offers additional advantages by promoting rapid tablet disintegration and faster drug release following administration. Such formulations are particularly beneficial in antidiabetic therapy, where consistent and predictable plasma drug concentrations are essential for maintaining optimal glycemic control. The application of statistical optimization tools, such as factorial design, and modern pharmaceutical development approaches, including Quality by Design (QbD), further facilitates the identification of critical formulation variables and ensures robust product performance throughout the product lifecycle. the development of improved formulation strategies for canagliflozin is essential to overcome its inherent biopharmaceutical limitations and to maximize its therapeutic potential in the treatment of Type 2 Diabetes Mellitus. The integration of solubility enhancement techniques with scientifically optimized formulation methodologies represents a promising approach for achieving improved oral bioavailability, enhanced clinical efficacy, and greater patient benefit.
2.DRUG PROFILE:
2.1 Canagliflozin:
Canagliflozin is an orally active antihyperglycemic agent belonging to the class of Sodium-Glucose Co-transporter-2 (SGLT2) inhibitors, which are widely used for the treatment of Type 2 Diabetes Mellitus (T2DM). It was one of the first SGLT2 inhibitors approved for clinical use and has significantly transformed diabetes management due to its unique insulin-independent mechanism of action and additional cardiovascular and renal protective benefits. Unlike conventional antidiabetic drugs such as sulfonylureas, which stimulate insulin secretion, or metformin, which primarily reduces hepatic glucose production, canagliflozin acts by targeting renal glucose handling. By inhibiting glucose reabsorption in the proximal renal tubules, it promotes urinary glucose excretion and lowers blood glucose levels without directly affecting insulin secretion or insulin sensitivity. This distinctive mechanism makes canagliflozin effective even in advanced stages of T2DM where pancreatic β-cell function may be compromised.
2.1.1 Chemical Information:
Fig.2. chemical parameters of Canagliflozin
|
Parameter |
Description |
|
Generic Name |
Canagliflozin |
|
IUPAC Name |
(2S,3R,4R,5S,6R)-2-[3-[[5-(4-fluorophenyl)thiophen-2-yl]methyl]-4-methylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol |
|
Molecular Formula |
C₂₄H₂₅FO₅S |
|
Molecular Weight |
444.52 g/mol |
|
Drug Category |
SGLT2 Inhibitor |
|
Therapeutic Class |
Oral Antidiabetic Agent |
|
Route of Administration |
Oral |
|
Dosage Forms Available |
Tablets |
|
BCS Classification |
Class IV Drug |
2.1.2.Chemical Structure:
Canagliflozin belongs to the C-glucoside derivative class, characterized by a glucose moiety covalently attached to a substituted aromatic ring system through a stable carbon-carbon bond.
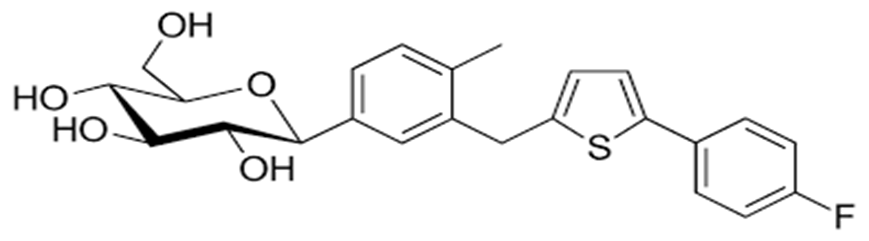
Fig.3 Structure of Canagliflozin
Chemically, canagliflozin is designated as (2S,3R,4R,5S,6R)-2-[3-[[5-(4- fluorophenyl) thiophen-2-yl]methyl]-4-methylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol, with a molecular formula of C₂₄H₂₅FO₅S and a molecular weight of 444.52 g/mol. The structure consists of three major structural domains, namely the β-D-glucopyranose moiety, the central aromatic phenyl ring, and the aryl-thiophene lipophilic region.
2.1.3 Mechanism of action:
Canagliflozin is a selective and reversible inhibitor of Sodium-Glucose Co-transporter-2 (SGLT2), an integral membrane protein predominantly expressed in the S1 segment of the proximal convoluted tubules of the kidneys. The primary physiological function of SGLT2 is to mediate the reabsorption of filtered glucose from the glomerular filtrate back into the systemic circulation, thereby maintaining glucose homeostasis under normal conditions. Approximately 180 g of glucose is filtered daily by the renal glomeruli, of which nearly 90% is reabsorbed through SGLT2 transporters, while the remaining 10% is reabsorbed by SGLT1 transporters located in the distal segments of the proximal tubule.
In patients with Type 2 Diabetes Mellitus (T2DM), the renal threshold for glucose reabsorption is often elevated due to increased expression and activity of SGLT2 transporters, resulting in excessive glucose retention and persistent hyperglycemia. This adaptive response contributes significantly to the maintenance and progression of elevated blood glucose concentrations in diabetic individuals. Canagliflozin exerts its therapeutic effect by selectively inhibiting SGLT2-mediated glucose transport, thereby reducing renal glucose reabsorption and increasing urinary glucose excretion (UGE). Consequently, excess glucose is eliminated through urine, leading to a reduction in both fasting and postprandial plasma glucose concentrations.
A distinctive feature of canagliflozin is that its antihyperglycemic activity is independent of pancreatic β-cell function and insulin secretion. Unlike conventional antidiabetic agents such as sulfonylureas or insulin secretagogues, canagliflozin does not stimulate insulin release and therefore carries a relatively low risk of hypoglycemia when administered as monotherapy. This insulin-independent mechanism enables effective glycemic control even in patients with advanced stages of T2DM, where progressive β-cell dysfunction may limit the efficacy of other therapeutic agents.
In addition to improving glycemic control, inhibition of renal glucose reabsorption results in the loss of calories through urinary glucose excretion, contributing to modest reductions in body weight and adiposity. Furthermore, the osmotic diuresis induced by glucosuria promotes mild natriuresis and reduction in intravascular volume, which may contribute to clinically significant reductions in systolic blood pressure. These additional metabolic and hemodynamic effects have expanded the therapeutic role of canagliflozin beyond glucose lowering alone. Clinical studies have further demonstrated that canagliflozin provides substantial cardiovascular and renal protective benefits, including reduction in the risk of hospitalization due to heart failure, slowing the progression of diabetic nephropathy, and decreasing albuminuria in patients with chronic kidney disease. These pleiotropic effects have established SGLT2 inhibitors as an important component of contemporary diabetes management guidelines, particularly for patients with established cardiovascular disease or renal impairment.
Despite its significant therapeutic advantages, the clinical performance of canagliflozin may be influenced by its unfavorable physicochemical properties, including poor aqueous solubility and limited oral bioavailability. Therefore, the development of advanced formulation approaches such as solid dispersion systems and optimized immediate-release formulations is essential to maximize drug dissolution, improve systemic exposure, and enhance therapeutic outcomes in patients with Type 2 Diabetes Mellitus.
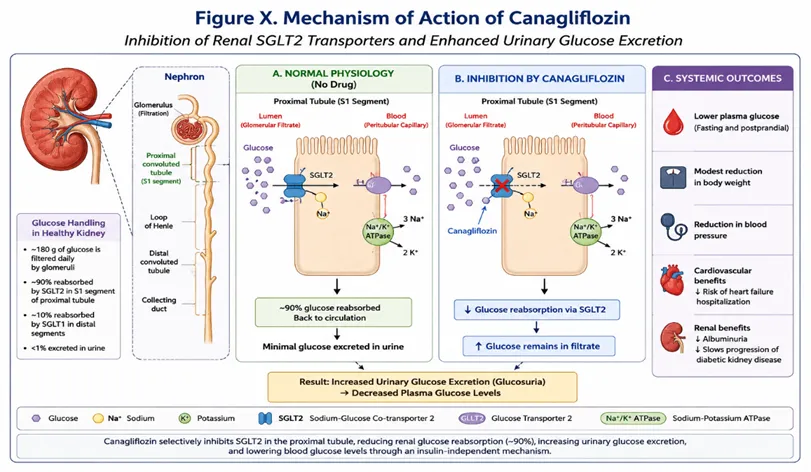
Fig.4. Mechanism of action of canagliflozin
2.1.4 PHARMACOKINETICS:
Canagliflozin, a selective sodium–glucose cotransporter-2 (SGLT2) inhibitor, exhibits pharmacokinetic characteristics that support its clinical utility as a once-daily oral antidiabetic agent. Following oral administration, the drug is rapidly absorbed from the gastrointestinal tract, with maximum plasma concentrations (CmaxC_{max}Cmax) generally achieved within 1–2 hours. Although canagliflozin demonstrates efficient gastrointestinal absorption, its absolute oral bioavailability is approximately 65%, primarily due to its poor aqueous solubility and the influence of first-pass metabolism. The absorption process is largely unaffected by food intake, although administration with high-fat meals may slightly delay the rate of absorption without significantly altering overall systemic exposure. These properties contribute to a predictable and dose-proportional pharmacokinetic profile over the therapeutic dosage range of 100–300 mg.
Once absorbed, canagliflozin exhibits extensive plasma protein binding, exceeding 99%, predominantly to serum albumin. Despite this high binding affinity, sufficient unbound drug remains available to exert pharmacological activity at renal SGLT2 transporters. The compound demonstrates moderate tissue distribution and maintains therapeutically effective plasma concentrations due to its relatively prolonged elimination half-life of approximately 10–13 hours, thereby enabling convenient once-daily administration and improving patient adherence. Metabolic elimination of canagliflozin occurs predominantly through phase II biotransformation pathways. The principal metabolic route involves O-glucuronidation mediated by uridine diphosphate glucuronosyltransferase enzymes, mainly UGT1A9 and UGT2B4, resulting in the formation of pharmacologically inactive metabolites. In contrast, cytochrome P450-mediated metabolism contributes minimally to its biotransformation, thereby reducing the likelihood of clinically significant CYP-associated drug–drug interactions.
Excretion of canagliflozin and its metabolites occurs through both renal and fecal pathways, with inactive glucuronide conjugates being eliminated primarily in urine and feces. However, renal impairment can significantly alter systemic drug exposure and diminish glucose-lowering efficacy because the therapeutic action of canagliflozin depends on adequate glomerular filtration and urinary glucose excretion. Consequently, dose adjustment and careful monitoring of renal function are recommended in patients with moderate to severe renal dysfunction. Overall, the pharmacokinetic profile of canagliflozin supports its effectiveness as a once-daily therapy while highlighting the importance of formulation strategies aimed at improving its solubility and bioavailability in oral dosage forms.
2.1.5. CLINICAL APPLICATIONS:
Canagliflozin has emerged as an important therapeutic agent in the management of type 2 diabetes mellitus (T2DM) owing to its insulin-independent mechanism of action and its favorable effects extending beyond glycemic control. Its primary clinical indication is the improvement of glycemic regulation in adult patients with T2DM, either as monotherapy in individuals intolerant to metformin or as part of combination therapy with other antidiabetic agents, including metformin, sulfonylureas, dipeptidyl peptidase-4 inhibitors, glucagon-like peptide-1 receptor agonists, and insulin. By selectively inhibiting renal sodium–glucose cotransporter-2 (SGLT2), canagliflozin promotes urinary glucose excretion, resulting in reductions in fasting plasma glucose concentrations and glycated hemoglobin (HbA1c) levels. Beyond glucose lowering, canagliflozin has demonstrated significant cardiovascular benefits in patients with established cardiovascular disease or those at high cardiovascular risk. Clinical outcome studies have reported reductions in major adverse cardiovascular events, including cardiovascular mortality, non-fatal myocardial infarction, and stroke. Furthermore, the drug has shown efficacy in decreasing the incidence of hospitalization due to heart failure, thereby expanding its role in cardiometabolic disease management.
Canagliflozin also possesses substantial renoprotective properties and is increasingly utilized in patients with diabetic kidney disease. By reducing intraglomerular pressure and restoring tubuloglomerular feedback mechanisms, it slows the progression of chronic kidney disease, decreases albuminuria, and lowers the risk of renal function decline and end-stage renal disease. These benefits are particularly valuable in patients with concomitant diabetes and renal impairment. Additionally, the drug contributes to modest reductions in body weight and blood pressure as a consequence of caloric loss through glucosuria and osmotic diuresis. These metabolic advantages make canagliflozin particularly suitable for overweight or obese patients with T2DM requiring comprehensive cardiometabolic risk reduction. Consequently, current clinical guidelines recommend canagliflozin not only for glycemic management but also for cardiovascular and renal risk mitigation in appropriately selected patients with type 2 diabetes mellitus.
3. BIOPHARMACEUTICAL CHALLENGES OF CANAGLIFLOZIN:
3.1. BCS CLASSIFICATION:
Canagliflozin is generally classified as a Biopharmaceutics Classification System (BCS) Class IV drug, characterized by low aqueous solubility and low intestinal permeability, which collectively limit its oral bioavailability and therapeutic performance. The poor solubility of canagliflozin in gastrointestinal fluids results in a dissolution rate-limited absorption process, while its relatively limited permeability across the intestinal epithelium further restricts systemic drug exposure. Consequently, conventional oral formulations may exhibit variable absorption profiles and inconsistent pharmacokinetic behavior.
The crystalline nature of canagliflozin further contributes to its poor dissolution characteristics by reducing the availability of drug molecules for absorption. These physicochemical limitations present significant formulation challenges and necessitate the application of advanced drug delivery strategies aimed at improving both solubility and dissolution rate. Approaches such as solid dispersion technology, amorphization, lipid-based delivery systems, nanocarriers, and immediate-release formulations incorporating hydrophilic excipients have been extensively investigated to enhance its oral performance.
From a pharmaceutical development perspective, the BCS Class IV designation emphasizes the importance of formulation optimization to achieve reproducible bioavailability and therapeutic efficacy. Improvement in dissolution behavior can substantially enhance systemic drug exposure, reduce interindividual variability, and ensure more consistent glycemic control in patients with type 2 diabetes mellitus. Therefore, overcoming the biopharmaceutical limitations associated with canagliflozin remains a critical objective in the design of efficient oral dosage forms.
Table 2. BSC CLASSIFICATION
|
BCS Class |
Solubility |
Permeability |
Rate-Limiting Step for Absorption |
Formulation Considerations |
|
Class I |
High |
High |
Gastric emptying |
Conventional immediate-release formulations are generally sufficient. |
|
Class II |
Low |
High |
Dissolution rate |
Solubility enhancement strategies such as solid dispersions and particle size reduction are often required. |
|
Class III |
High |
Low |
Membrane permeability |
Permeability enhancement approaches and excipient optimization are necessary. |
|
Class IV |
Low |
Low |
Both dissolution and permeability limitations |
Advanced formulation strategies including solid dispersions, nanocarriers, lipid-based systems, and amorphous formulations are generally required to improve oral bioavailability. |
3.2. POOR AQUEOUS SOLUBILITY:
One of the major pharmaceutical limitations of canagliflozin is its poor aqueous solubility, which significantly influences its dissolution behavior and oral bioavailability. The drug exists predominantly in a highly crystalline form, resulting in strong intermolecular interactions within the crystal lattice that hinder its dissolution in gastrointestinal fluids. Consequently, only a limited fraction of the administered dose becomes available in dissolved form for absorption across the intestinal membrane, making dissolution the rate-limiting step in its oral absorption process. The low aqueous solubility of canagliflozin contributes to variability in plasma drug concentrations and may lead to inconsistent therapeutic responses among patients. Furthermore, physiological factors such as gastrointestinal pH, gastric emptying time, intestinal motility, and fed or fasted conditions can further influence its dissolution characteristics and subsequent absorption. These challenges are particularly relevant in oral immediate-release formulations, where rapid dissolution is essential for achieving prompt onset of antihyperglycemic activity.
From a formulation perspective, overcoming the solubility limitations of canagliflozin remains a critical objective in pharmaceutical development. Various solubility enhancement strategies, including solid dispersion technology, amorphization, particle size reduction, lipid-based delivery systems, nanocarriers, and the incorporation of hydrophilic polymers, have been extensively investigated to improve dissolution kinetics and enhance systemic drug exposure. Conversion of the crystalline drug into an amorphous form has been reported to substantially increase solubility and dissolution rates by reducing crystal lattice energy and increasing molecular mobility.
Improvement of the aqueous solubility of canagliflozin is therefore essential for achieving consistent bioavailability, predictable pharmacokinetic behavior, and reliable therapeutic efficacy in the management of type 2 diabetes mellitus. The development of optimized formulation approaches aimed at enhancing dissolution performance remains a key strategy for maximizing the clinical potential of this SGLT2 inhibitor.
3.3. LOW DISSOLUTION RATE OF CANAGLIFLOZIN:
The low dissolution rate of canagliflozin represents a major challenge in its oral formulation development and is primarily attributed to its poor aqueous solubility and highly crystalline nature. Since dissolution is a prerequisite for gastrointestinal absorption, the slow dissolution of canagliflozin in biological fluids can delay drug absorption and limit the amount of drug available for systemic circulation. Consequently, the dissolution process becomes a critical determinant of its pharmacokinetic performance and therapeutic efficacy.
The crystalline structure of canagliflozin possesses high lattice energy, which restricts the release of drug molecules into the dissolution medium. As a result, conventional oral formulations often exhibit incomplete or delayed drug dissolution, potentially leading to variable plasma concentrations and inconsistent glycemic control among patients with type 2 diabetes mellitus. Furthermore, physiological factors such as gastrointestinal pH, motility, and food intake may further influence dissolution behavior and contribute to interindividual variability in drug absorption.From a biopharmaceutical perspective, the dissolution rate of canagliflozin constitutes the principal rate-limiting step governing its oral absorption. Therefore, improving dissolution characteristics is essential to achieve rapid onset of action, enhanced bioavailability, and predictable therapeutic outcomes. Numerous formulation strategies have been investigated to overcome this limitation, including particle size reduction, solid dispersion systems, amorphization, lipid-based formulations, nanosuspensions, and the incorporation of hydrophilic carriers and superdisintegrants.Among these approaches, solid dispersion technology has demonstrated particular promise by converting the crystalline drug into an amorphous or molecularly dispersed state, thereby increasing the effective surface area available for dissolution and reducing crystal lattice energy. Such modifications significantly improve dissolution kinetics and facilitate more rapid drug absorption following oral administration.Therefore, enhancement of the dissolution rate remains a fundamental objective in the formulation development of canagliflozin to ensure improved oral bioavailability, reduced pharmacokinetic variability, and consistent therapeutic efficacy in the management of type 2 diabetes mellitus.
3.4. Bioavailability Limitations of Canagliflozin:
The oral bioavailability of canagliflozin is constrained by several physicochemical and biopharmaceutical factors that collectively limit its therapeutic performance. One of the principal limitations is its poor aqueous solubility, which restricts the amount of drug available in dissolved form for absorption across the gastrointestinal epithelium. Since dissolution is a prerequisite for absorption, inadequate solubilization in gastrointestinal fluids results in incomplete and variable systemic drug exposure.
In addition to poor solubility, the crystalline nature of canagliflozin contributes to its slow dissolution kinetics by increasing crystal lattice energy and reducing molecular mobility. This characteristic further impedes drug release from conventional oral dosage forms and delays the attainment of therapeutic plasma concentrations. Variability in gastrointestinal conditions, including pH, gastric emptying time, intestinal motility, and fed or fasted states, may further influence dissolution and absorption, leading to inter- and intra-subject variability in pharmacokinetic behavior. Another important factor limiting bioavailability is hepatic first-pass metabolism, which reduces the fraction of the orally administered dose reaching the systemic circulation in an unchanged form. Although canagliflozin exhibits efficient gastrointestinal absorption, metabolic conversion during its first passage through the liver decreases overall systemic availability. Consequently, the absolute oral bioavailability of crystalline canagliflozin has been reported to be approximately 65%.
These biopharmaceutical limitations can result in fluctuations in plasma drug concentrations and may compromise the consistency of glycemic control in patients with type 2 diabetes mellitus. Therefore, the development of advanced formulation approaches aimed at improving solubility, dissolution rate, and systemic exposure has become an important area of pharmaceutical research. Strategies such as solid dispersion systems, amorphous formulations, lipid-based carriers, nanosuspensions, and hydrophilic polymer incorporation have demonstrated considerable potential in overcoming these limitations and enhancing oral bioavailability.
Overall, addressing the bioavailability challenges associated with canagliflozin is essential for achieving predictable pharmacokinetic performance, improved therapeutic efficacy, and consistent clinical outcomes in the treatment of type 2 diabetes mellitus.
Table 3. Bioavailability Limitations of Canagliflozin
|
Limitation |
Impact on Drug Performance |
|
Poor aqueous solubility |
Reduces the amount of dissolved drug available for absorption |
|
Low dissolution rate |
Delays drug release and gastrointestinal absorption |
|
Crystalline structure |
Increases lattice energy and limits dissolution |
|
First-pass metabolism |
Decreases the fraction of drug reaching systemic circulation |
|
Gastrointestinal variability |
Causes inter- and intra-patient variability in absorption |
|
BCS Class IV characteristics |
Results in both solubility- and permeability-related absorption challenges |
|
Variable systemic exposure |
May contribute to inconsistent therapeutic response |
3.5. STABILITY ISSUES:
The pharmaceutical development of canagliflozin is significantly influenced by its stability-related challenges, particularly when employing solubility enhancement techniques such as solid dispersions and amorphous formulations. Although these approaches improve dissolution characteristics and bioavailability, they may compromise the physical and chemical stability of the drug during storage and processing.
A major stability concern is the tendency of amorphous or molecularly dispersed canagliflozin to undergo recrystallization over time. Since the amorphous form exists in a thermodynamically unstable high-energy state, it may gradually revert to its more stable crystalline form during storage, especially under conditions of elevated temperature and humidity. Such recrystallization can significantly reduce dissolution rate and subsequently decrease oral bioavailability and therapeutic efficacy. Canagliflozin formulations may also exhibit sensitivity to environmental factors, including moisture, temperature fluctuations, and light exposure, which can adversely affect the physicochemical properties of the dosage form. Moisture uptake may alter powder flow characteristics, promote agglomeration, and accelerate degradation processes, while elevated temperatures may increase molecular mobility and facilitate crystallization within solid dispersion systems.
Another important consideration is the drug–excipient compatibility within the formulation matrix. Inappropriate selection of carriers, polymers, or excipients may result in chemical interactions that adversely influence drug stability, dissolution behavior, or content uniformity during long-term storage. Therefore, compatibility studies using analytical techniques such as Fourier-transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), and X-ray diffraction (XRD) are essential during formulation development. To mitigate these stability concerns, careful optimization of formulation composition, manufacturing conditions, and packaging systems is required. The use of suitable stabilizing polymers, moisture-protective packaging materials, and controlled storage conditions can help preserve the amorphous state and maintain the desired dissolution profile throughout the product shelf life.
Overall, addressing stability-related challenges is crucial for ensuring the long-term quality, efficacy, and reproducibility of canagliflozin formulations, particularly those utilizing advanced drug delivery technologies intended to enhance oral bioavailability.
4. SOLUBILITY ENHANCEMENT STRATEGIES FOR CANAGLIFLOZIN:
The poor aqueous solubility of canagliflozin represents a major limitation to its oral bioavailability and therapeutic performance. As a poorly water-soluble drug, canagliflozin exhibits dissolution-limited absorption, necessitating the application of advanced formulation approaches to improve its solubility and dissolution characteristics. Various pharmaceutical strategies have been investigated to enhance its oral absorption and ensure consistent therapeutic efficacy.
4.1. PARTICLE SIZE REDUCTION AS A SOLUBILITY ENHANCEMENT STRATEGY FOR CANAGLIFLOZIN:
Particle size reduction is a widely employed pharmaceutical strategy to enhance the dissolution characteristics and oral bioavailability of poorly water-soluble drugs such as canagliflozin. The principle underlying this approach is based on increasing the surface area available for interaction with the dissolution medium. According to the Noyes–Whitney equation, a reduction in particle size results in an increase in surface area, thereby accelerating the dissolution rate and improving drug absorption from the gastrointestinal tract. Canagliflozin exhibits poor aqueous solubility and slow dissolution owing to its crystalline structure and high lattice energy. Consequently, reducing the particle size to the micron or nanometer range can significantly improve wetting properties, increase saturation solubility, and facilitate rapid dissolution in gastrointestinal fluids. Enhanced dissolution ultimately contributes to improved systemic drug exposure and more consistent therapeutic efficacy.
Several particle size reduction techniques have been investigated for poorly soluble compounds, including micronization, nanonization, wet milling, high-pressure homogenization, and nanosuspension technology. Among these, nanocrystal and nanosuspension formulations have shown particular promise because they not only increase the surface area but also improve adhesion to the gastrointestinal mucosa, thereby prolonging residence time and enhancing absorption. Despite its advantages, particle size reduction may present certain challenges, including particle aggregation, poor flowability, increased surface energy, and physical instability during storage. Therefore, stabilizers and surfactants are often incorporated into the formulation to maintain particle dispersion and prevent agglomeration. In the case of canagliflozin, particle size reduction is frequently combined with other formulation strategies such as solid dispersion technology, lipid-based carriers, or hydrophilic polymers to achieve synergistic improvements in dissolution and bioavailability.
Overall, particle size reduction represents an effective and practical approach for overcoming the dissolution-limited absorption of canagliflozin and contributes significantly to the development of efficient oral dosage forms for the management of type 2 diabetes mellitus.
4.2. MICRONIZATION AND NANONIZATION OF CANAGLIFLOZIN:
Micronization and nanonization are well-established particle engineering approaches employed to improve the dissolution characteristics and oral bioavailability of poorly water-soluble drugs such as canagliflozin. Both techniques operate by reducing particle size and consequently increasing the effective surface area available for interaction with gastrointestinal fluids, thereby enhancing dissolution kinetics according to the Noyes–Whitney equation.
Micronization involves reducing drug particles to the micrometer range, typically between 1 and 10 µm, using techniques such as jet milling, ball milling, or fluid energy milling. The resulting increase in surface area improves wettability and accelerates dissolution, thereby enhancing drug absorption following oral administration. However, micronized particles may exhibit poor flow properties and a tendency toward aggregation due to increased surface energy, potentially limiting their formulation performance. In contrast, nanonization reduces particle dimensions to the nanometer scale, generally below 1000 nm. Nanocrystals and nanosuspensions produced through high-pressure homogenization, wet milling, or precipitation techniques exhibit substantially greater surface area and saturation solubility compared with micronized particles. Furthermore, nanosized particles may improve adhesion to the intestinal mucosa and prolong gastrointestinal residence time, thereby facilitating enhanced absorption and increased systemic exposure.
For canagliflozin, nanonization offers several advantages over conventional micronization because the drug's poor aqueous solubility represents a major barrier to efficient oral delivery. The reduction of particle size to the nanoscale not only enhances dissolution velocity but may also improve apparent solubility and reduce pharmacokinetic variability. Nevertheless, nanosystems often require stabilizers surfactants to prevent particle aggregation and maintain long-term physical stability during storage. Consequently, both micronization and nanonization represent valuable formulation strategies for overcoming dissolution-related limitations associated with canagliflozin. However, nanonization is generally considered the more effective approach due to its superior impact on solubility enhancement and bioavailability improvement.
4.3CYCLODEXTRIN COMPLEXATION AS A SOLUBILITY ENHANCEMENT STRATEGY FOR CANAGLIFLOZIN:
Cyclodextrin complexation is an effective pharmaceutical approach for improving the aqueous solubility and dissolution characteristics of poorly water-soluble drugs such as canagliflozin. Cyclodextrins are cyclic oligosaccharides composed of α-(1,4)-linked glucopyranose units that possess a unique molecular architecture characterized by a hydrophobic internal cavity and a hydrophilic external surface. This structural arrangement enables cyclodextrins to form non-covalent inclusion complexes with lipophilic drug molecules, thereby enhancing their apparent aqueous solubility and dissolution behavior.
In the case of canagliflozin, the hydrophobic aromatic moieties of the molecule can be partially accommodated within the hydrophobic cavity of cyclodextrins, while the hydrophilic outer surface of the complex remains exposed to the aqueous environment. This molecular encapsulation improves drug wettability, reduces particle aggregation, and increases the concentration of dissolved drug available for absorption in the gastrointestinal tract. Consequently, cyclodextrin complexation can significantly enhance dissolution rate and oral bioavailability. Among the various cyclodextrin derivatives, β-cyclodextrin (β-CD) and its modified forms, including hydroxypropyl-β-cyclodextrin (HP-β-CD) and sulfobutyl ether-β-cyclodextrin (SBE-β-CD), are most commonly employed due to their superior aqueous solubility and favorable safety profiles. These derivatives exhibit greater complexation efficiency and improved pharmaceutical applicability compared with native cyclodextrins.
The inclusion complexes can be prepared using several techniques, including kneading, co-precipitation, solvent evaporation, freeze-drying, and spray-drying methods. The selection of preparation method influences complexation efficiency, particle characteristics, and dissolution performance of the final product. Despite its advantages, cyclodextrin complexation may be associated with certain limitations, including limited drug-loading capacity, increased formulation cost, and potential stability issues at high humidity conditions. Nevertheless, it remains a promising strategy for improving the biopharmaceutical performance of canagliflozin and other BCS Class IV compounds.
4.4. NANOCRYSTALS AND NANOSUSPENSIONS AS SOLUBILITY ENHANCEMENT STRATEGIES FOR CANAGLIFLOZIN:
Nanocrystal and nanosuspension technologies have emerged as promising nanotechnological approaches for improving the solubility, dissolution rate, and oral bioavailability of poorly water-soluble drugs such as canagliflozin. Owing to its low aqueous solubility and BCS Class IV characteristics, canagliflozin exhibits dissolution-limited absorption, necessitating advanced formulation strategies capable of enhancing its biopharmaceutical performance.
Nanocrystals are submicron-sized particles composed almost entirely of the pure drug substance, typically ranging from 100 to 1000 nm in diameter and stabilized by surfactants or polymers to prevent aggregation. Reduction of particle size to the nanometer scale substantially increases the surface area available for dissolution, resulting in enhanced saturation solubility and accelerated dissolution kinetics. According to the Ostwald–Freundlich relationship, the increased curvature of nanoparticles contributes to an elevation in apparent solubility, thereby improving gastrointestinal absorption and systemic drug exposure. Nanocrystals are commonly prepared using top-down techniques such as wet media milling and high-pressure homogenization. In contrast, nanosuspensions consist of finely dispersed drug nanoparticles suspended in an aqueous medium and stabilized by surfactants, polymers, or dispersing agents. Similar to nanocrystals, nanosuspensions provide a significant increase in effective surface area and dissolution velocity; however, the presence of a stabilizing liquid medium facilitates uniform particle dispersion and minimizes agglomeration. Furthermore, nanosuspensions may enhance mucosal adhesion and prolong gastrointestinal residence time, thereby improving intestinal absorption and reducing pharmacokinetic variability. Both approaches offer several advantages, including improved wettability, enhanced dissolution rate, increased oral bioavailability, and the potential for dose reduction. Nevertheless, challenges such as physical instability, particle aggregation, and Ostwald ripening may compromise long-term storage stability and therefore require the incorporation of suitable stabilizers.
Overall, nanocrystal and nanosuspension formulations represent highly effective strategies for overcoming the solubility and dissolution limitations associated with canagliflozin, thereby improving its therapeutic performance and clinical efficacy in the management of type 2 diabetes mellitus.
4.4. CO-CRYSTALS AS A SOLUBILITY ENHANCEMENT STRATEGY FOR CANAGLIFLOZIN:
Pharmaceutical co-crystallization has emerged as an effective crystal engineering approach for improving the solubility and dissolution characteristics of poorly water-soluble drugs such as canagliflozin. A pharmaceutical co-crystal is a crystalline material composed of an active pharmaceutical ingredient (API) and one or more pharmaceutically acceptable co-formers held together by non-covalent intermolecular interactions, primarily hydrogen bonding, π–π stacking, and van der Waals forces. Unlike salts, co-crystals do not involve proton transfer and therefore preserve the molecular integrity and pharmacological activity of the parent drug. The poor aqueous solubility and slow dissolution rate of canagliflozin are largely attributed to its highly ordered crystalline lattice and strong intermolecular interactions. Co-crystal formation modifies the crystal packing arrangement and reduces lattice energy, thereby facilitating greater interaction between drug molecules and the dissolution medium. Consequently, co-crystals exhibit enhanced wettability, improved dissolution kinetics, and increased apparent solubility, leading to improved oral absorption and systemic exposure.
Selection of an appropriate co-former is critical for successful co-crystal development. Commonly employed co-formers include pharmaceutically acceptable compounds such as nicotinamide, saccharin, fumaric acid, succinic acid, citric acid, and malonic acid, which possess functional groups capable of establishing stable intermolecular interactions with the drug molecule. Co-crystals can be prepared using several techniques, including solvent evaporation, slurry conversion, grinding, spray drying, and solvent-drop grinding methods.
In addition to enhancing solubility, co-crystal technology may improve physicochemical properties such as flowability, compressibility, hygroscopicity, and thermal stability without altering the pharmacological properties of the active ingredient. However, challenges including polymorphic transitions, scale-up difficulties, and long-term physical stability must be carefully addressed during formulation development.
5. SOLID DISPERSION TECHNOLOGY:
In a solid dispersion system, the drug may exist in an amorphous state, as molecular dispersions, or as extremely fine crystalline particles within the polymeric carrier. Conversion of canagliflozin from its stable crystalline form to a metastable amorphous form reduces crystal lattice energy and enhances molecular mobility, resulting in significantly improved dissolution kinetics and apparent solubility. Furthermore, hydrophilic carriers facilitate rapid penetration of dissolution media and promote uniform drug dispersion, thereby increasing the concentration gradient required for intestinal absorption.
5.1. DEFINITION AND CONCEPT:
Solid dispersion technology is defined as a pharmaceutical formulation approach in which one or more active pharmaceutical ingredients (APIs) are dispersed within an inert carrier or matrix in the solid state to improve their physicochemical and biopharmaceutical properties, particularly aqueous solubility and dissolution rate. The drug may be present in an amorphous form, as molecularly dispersed entities, or as fine crystalline particles embedded within a hydrophilic carrier matrix. This technique is especially beneficial for poorly water-soluble compounds belonging to BCS Class II and Class IV categories, where dissolution represents the primary barrier to oral absorption.
CONCEPT: The fundamental concept of solid dispersion technology is based on enhancing the dissolution behavior of poorly soluble drugs by reducing particle size to the molecular level, improving wettability, increasing surface area, and converting the crystalline drug into a less ordered amorphous state. The incorporation of hydrophilic carriers facilitates rapid penetration of dissolution media and promotes efficient drug dispersion within the gastrointestinal environment, thereby increasing the concentration of dissolved drug available for absorption. For canagliflozin, the poor aqueous solubility and highly crystalline nature significantly limit its dissolution and oral bioavailability. Solid dispersion systems overcome these limitations by dispersing the drug within hydrophilic polymers such as polyvinylpyrrolidone (PVP K30) and polyethylene glycol (PEG 6000), resulting in reduced crystallinity and enhanced molecular mobility. Consequently, the dissolution rate and apparent solubility of canagliflozin are markedly improved, leading to enhanced systemic exposure and therapeutic efficacy. The mechanisms responsible for solubility enhancement in solid dispersions include particle size reduction, amorphization, improved wettability, enhanced porosity, and prevention of particle aggregation. Collectively, these effects contribute to faster dissolution and more predictable absorption following oral administration.
Therefore, solid dispersion technology represents a highly effective and versatile strategy for overcoming the biopharmaceutical limitations of poorly soluble drugs and has become one of the most extensively investigated approaches for improving the oral delivery of canagliflozin.
5.2. CLASSIFICATION OF SOLID DISPERSIONS:
Solid dispersions are commonly classified into four generations based on the nature of the carrier employed and the mechanism by which solubility enhancement is achieved. The evolution from first- to fourth-generation systems reflects progressive improvements in dissolution enhancement, physical stability, and bioavailability of poorly water-soluble drugs.
5..2.1. First-Generation Solid Dispersions: First-generation solid dispersions utilize crystalline carriers to improve drug dissolution. These systems generally consist of eutectic mixtures in which both the drug and carrier remain in a crystalline state after solidification. Although particle size reduction resulting from the eutectic structure can improve dissolution rates, the enhancement in solubility is often limited due to the persistence of crystallinity. Common carriers employed in first-generation systems include urea, mannitol, sorbitol, and succinic acid. These formulations exhibit relatively good physical stability but provide only modest improvements in dissolution compared with later-generation systems.
5.2.2. Second-Generation Solid Dispersions: Second-generation solid dispersions incorporate amorphous hydrophilic polymeric carriers, resulting in the conversion of crystalline drug molecules into an amorphous or molecularly dispersed state. Reduction of crystallinity and increased wettability contribute significantly to enhanced dissolution and bioavailability. Commonly used carriers include polyvinylpyrrolidone (PVP), polyethylene glycol (PEG), hydroxypropyl methylcellulose (HPMC), and polyethylene oxide (PEO). Because canagliflozin solid dispersions prepared using PVP K30 and PEG 6000 fall into this category, second-generation systems are particularly relevant for its formulation development.
5.2.3. Third-Generation Solid Dispersions: Third-generation solid dispersions employ surface-active agents and self-emulsifying carriers in addition to hydrophilic polymers. The incorporation of surfactants improves drug wettability, inhibits recrystallization, and enhances physical stability of the amorphous drug form. Examples of third-generation carriers include poloxamers, Gelucire®, Soluplus®, Tween 80, and sodium lauryl sulfate (SLS). These systems often provide superior dissolution enhancement compared with second-generation dispersions due to synergistic effects between polymers and surfactants.
5.2.4. Fourth-Generation Solid Dispersions: Fourth-generation solid dispersions are designed to provide controlled, sustained, or targeted drug release in addition to solubility enhancement. These advanced systems combine solubility improvement with modulation of drug release kinetics to optimize therapeutic performance. Carriers employed in fourth-generation systems include pH-responsive polymers, biodegradable polymers, and controlled-release matrices, such as Eudragit®, ethyl cellulose, and poly(lactic-co-glycolic acid) (PLGA). These formulations are particularly useful for drugs requiring prolonged therapeutic action or site-specific delivery.
6. MECHANISM OF SOLUBILITY ENHANCEMENT:
Solid dispersion technology is one of the most effective formulation strategies employed to improve the aqueous solubility and dissolution behavior of poorly water-soluble drugs. The enhancement in drug dissolution is attributed to several physicochemical modifications that occur when the active pharmaceutical ingredient (API) is molecularly dispersed within a hydrophilic carrier matrix. These modifications collectively improve drug wettability, decrease crystallinity, increase porosity, reduce particle size, and stabilize the amorphous form of the drug. The major mechanisms responsible for solubility enhancement are described below.
6.1.AMORPHIZATION:
Amorphization is considered one of the most significant mechanisms responsible for improving the solubility of poorly water-soluble drugs incorporated into solid dispersions. In the crystalline state, drug molecules are arranged in a highly ordered lattice stabilized by strong intermolecular interactions. Before dissolution can occur, these crystal lattice forces must be disrupted, requiring a considerable amount of energy and consequently limiting the dissolution rate. During the preparation of solid dispersions, the crystalline drug is converted partially or completely into an amorphous form, where molecules are randomly arranged without long-range structural order. Because the amorphous state possesses higher internal energy, increased molecular mobility, and greater Gibbs free energy than its crystalline counterpart, drug molecules dissolve more readily in aqueous media. The absence of crystal lattice energy significantly lowers the thermodynamic barrier to dissolution, thereby increasing both apparent solubility and dissolution rate.
Hydrophilic polymeric carriers, including polyvinylpyrrolidone (PVP), polyethylene glycol (PEG), hydroxypropyl methylcellulose (HPMC), and poloxamers, play an essential role in maintaining the amorphous state. These carriers interact with drug molecules through hydrogen bonding, dipole–dipole interactions, and van der Waals forces, thereby inhibiting molecular mobility and preventing recrystallization during storage. Consequently, amorphous solid dispersions exhibit improved physical stability together with enhanced dissolution performance, ultimately contributing to greater oral bioavailability.
6.2. WETTABILITY IMPROVEMENT:
Poor wettability is a major factor limiting the dissolution of hydrophobic drugs because insufficient contact between the drug surface and the dissolution medium delays water penetration and drug dissolution. Solid dispersion systems effectively overcome this limitation by incorporating hydrophilic carriers that readily absorb water and facilitate rapid hydration of the drug particles. When exposed to gastrointestinal fluids, hydrophilic polymers rapidly hydrate and form a thin aqueous layer surrounding the dispersed drug particles. This process decreases the interfacial tension between the hydrophobic drug surface and the dissolution medium, promoting rapid wetting and increasing the effective surface available for dissolution. Improved wettability also minimizes particle aggregation, ensuring uniform dispersion of drug particles throughout the dissolution medium.
Furthermore, certain carriers exhibit surfactant-like properties that enhance solvent penetration into the drug matrix and facilitate molecular dispersion of the API. The combined effects of improved hydration, enhanced solvent accessibility, and reduced interfacial resistance accelerate drug dissolution and significantly improve dissolution efficiency.
6.3. PARTICLE SIZE REDUCTION:
Reduction of particle size is another important mechanism responsible for the enhanced dissolution characteristics observed in solid dispersion systems. According to the Noyes–Whitney equation, the dissolution rate of a solid drug is directly proportional to its available surface area. Therefore, decreasing particle size substantially increases the surface exposed to the dissolution medium, leading to faster drug dissolution. In solid dispersions, the drug is often dispersed at the molecular, colloidal, or nanometer scale within the hydrophilic carrier matrix. This extensive reduction in particle dimensions dramatically increases the surface area available for solvent interaction compared with conventional crystalline drug particles. The homogeneous molecular distribution of the drug within the carrier also prevents particle agglomeration, ensuring maximum exposure to the dissolution medium.
Additionally, reduced diffusion distance enables rapid solvent penetration and facilitates immediate release of dissolved drug molecules into the surrounding medium. Consequently, particle size reduction significantly accelerates dissolution kinetics and enhances the bioavailability of poorly water-soluble pharmaceutical compounds.
6.4. POROSITY ENHANCEMENT:
The preparation of solid dispersions frequently results in the formation of porous particles possessing a large internal surface area. Increased porosity enhances the penetration of dissolution media into the interior of the drug matrix, thereby accelerating hydration, disintegration, and drug release.
The porous architecture facilitates rapid capillary action, allowing dissolution fluid to diffuse efficiently throughout the formulation. This increased fluid accessibility promotes faster swelling of hydrophilic carriers and enables immediate exposure of embedded drug molecules to the surrounding aqueous environment. As a result, dissolution occurs simultaneously throughout the porous network rather than being limited to the external particle surface. Moreover, the increased surface roughness and interconnected pore structure reduce diffusion resistance, allowing dissolved drug molecules to diffuse rapidly into the bulk dissolution medium. Consequently, enhanced porosity contributes substantially to improved dissolution kinetics, particularly for poorly water-soluble drugs formulated as solid dispersions.
6. POLYMERS AND CARRIERS USED IN CANAGLIFLOZIN SOLID DISPERSIONS:
Canagliflozin is a poorly water-soluble antidiabetic drug whose oral absorption is primarily limited by its low aqueous solubility and slow dissolution rate. As a result, solid dispersion technology has emerged as an effective formulation strategy to enhance its physicochemical and biopharmaceutical properties. The selection of an appropriate polymer or carrier is a critical determinant of the performance of solid dispersions, as these excipients influence drug solubilization, amorphization, wettability, dissolution rate, physical stability, and ultimately oral bioavailability. Hydrophilic polymers and amphiphilic carriers improve drug dispersion at the molecular level while simultaneously preventing recrystallization of the amorphous drug during storage. Various polymers have been investigated for the formulation of canagliflozin solid dispersions, each possessing distinct physicochemical characteristics and mechanisms of action
Table 4. Polymers and carriers
|
Polymer/Carrier |
Major Mechanism |
Major Advantages |
Common Preparation Methods |
|
PVP K30 |
Amorphization and hydrogen bonding |
Excellent dissolution enhancement and physical stability |
Spray drying, solvent evaporation |
|
PEG 4000/6000 |
Wettability improvement and particle dispersion |
Rapid dissolution and ease of processing |
Fusion method, solvent evaporation |
|
HPMC |
Crystallization inhibition |
Maintains supersaturation and prevents recrystallization |
Spray drying, solvent evaporation |
|
Poloxamer 188/407 |
Micellar solubilization and surfactant action |
Improved wettability and solubilization |
Fusion method, hot-melt extrusion |
|
Soluplus® |
Polymeric micelle formation |
High bioavailability and amorphous stability |
Spray drying, hot-melt extrusion |
|
Copovidone |
Molecular dispersion and hydrogen bonding |
Excellent stability and dissolution enhancement |
Spray drying, solvent evaporation |
7. CHARACTERIZATION TECHNIQUES:
Characterization of solid dispersions is essential to evaluate their physicochemical properties, confirm the successful incorporation of the drug into the carrier matrix, and assess their influence on solubility and dissolution behavior. Various analytical techniques are employed to investigate drug–polymer interactions, crystallinity, surface morphology, thermal behavior, and dissolution performance. These characterization methods provide comprehensive information regarding the stability, molecular dispersion, and pharmaceutical performance of solid dispersion systems.
7.1. FOURIER TRANSFORM INFRARED SPECTROSCOPY (FTIR):
Fourier Transform Infrared Spectroscopy (FTIR) is widely used to investigate possible interactions between the drug and polymer in solid dispersion formulations. The technique measures the absorption of infrared radiation by different functional groups present in the sample, producing a characteristic spectrum. Any shift, disappearance, or broadening of characteristic absorption peaks indicates hydrogen bonding or other intermolecular interactions between the drug and carrier. FTIR analysis also confirms the chemical compatibility of formulation components and verifies that no undesirable chemical degradation occurs during the preparation process.
7.2. DIFFERENTIAL SCANNING CALORIMETRY (DSC):
Differential Scanning Calorimetry (DSC) is a thermal analytical technique used to evaluate the thermal behavior of solid dispersions. It measures heat flow associated with physical transitions such as melting, crystallization, and glass transition. The disappearance or reduction of the drug's melting endotherm indicates successful conversion from the crystalline to the amorphous state. DSC also provides valuable information regarding drug-polymer miscibility, thermal stability, and possible interactions within the formulation.
7.3. SCANNING ELECTRON MICROSCOPY (SEM):
Scanning Electron Microscopy (SEM) is employed to examine the surface morphology and particle characteristics of solid dispersions. High-resolution images generated by SEM reveal particle shape, size, surface texture, and porosity. The technique also helps identify morphological changes following solid dispersion preparation, such as transformation from large crystalline particles to smooth, irregular, or porous amorphous structures. These changes are often associated with enhanced dissolution properties.
8. QUALITY BY DESIGN (QBD) AND FACTORIAL DESIGN:
The formulation of canagliflozin solid dispersions requires systematic optimization to achieve enhanced solubility, rapid dissolution, and improved oral bioavailability. Conventional formulation development often relies on trial-and-error experimentation, which is time-consuming and may not adequately explain the influence of formulation variables on product performance. To overcome these limitations, the Quality by Design (QbD) approach has become an integral part of modern pharmaceutical development. QbD is a science-based, risk-oriented strategy that emphasizes designing product quality during development rather than relying solely on final product testing. According to the International Council for Harmonisation (ICH Q8, Q9, and Q10), QbD involves understanding the relationship between material attributes, manufacturing processes, and product performance to ensure consistent quality throughout the product lifecycle.
In the development of canagliflozin solid dispersions, QbD facilitates the systematic selection of polymers, optimization of drug-to-polymer ratio, preparation method, and processing conditions. Statistical Design of Experiments (DoE) tools, including factorial design and response surface methodology, are employed to evaluate the influence of formulation variables on critical quality attributes such as solubility, dissolution rate, amorphous conversion, and physical stability. This systematic methodology minimizes experimental trials while improving process understanding and formulation robustness.
8.1. QUALITY BY DESIGN (QBD) PRINCIPLES:
he first step in QbD is the establishment of the Quality Target Product Profile (QTPP), which defines the desired characteristics of the final dosage form. For canagliflozin solid dispersions, the QTPP includes immediate drug release, enhanced aqueous solubility, improved dissolution rate, adequate stability, acceptable tablet properties, and increased oral bioavailability.
After defining the QTPP, formulation variables and process parameters that may influence product quality are identified through scientific knowledge and risk assessment. Hydrophilic carriers such as PVP K30 and PEG 6000, drug-polymer ratio, preparation technique, drying conditions, and mixing time are recognized as important variables affecting formulation performance. Risk assessment tools are subsequently employed to identify high-risk variables requiring further optimization. The identified variables are then investigated using statistical experimental designs, allowing researchers to establish mathematical relationships between formulation factors and pharmaceutical responses. Finally, an optimized design space is developed in which consistent product quality can be achieved with minimal variability. The application of QbD therefore provides scientific understanding, regulatory flexibility, and improved manufacturing reproducibility for canagliflozin solid dispersion formulations.
8.2. CRITICAL QUALITY ATTRIBUTES (CQAS):
Critical Quality Attributes are measurable physicochemical characteristics that directly influence the quality, safety, and therapeutic performance of pharmaceutical products. For canagliflozin solid dispersions, several CQAs are essential to ensure enhanced dissolution and bioavailability. Among these attributes, aqueous solubility and dissolution rate are considered the most critical because canagliflozin belongs to the Biopharmaceutics Classification System (BCS) Class IV and exhibits poor water solubility. The conversion of crystalline drug into an amorphous state is another important quality attribute since amorphization significantly improves dissolution characteristics. Uniform drug content ensures dose accuracy, while moisture content influences the physical stability of amorphous dispersions. Additional CQAs include particle size distribution, tablet hardness, friability, disintegration time, and long-term stability. Analytical techniques such as Fourier Transform Infrared Spectroscopy (FTIR), Differential Scanning Calorimetry (DSC), Powder X-ray Diffraction (PXRD), Scanning Electron Microscopy (SEM), and dissolution testing are commonly employed to evaluate these quality attributes. Maintaining these parameters within predetermined specifications ensures consistent formulation quality and therapeutic efficacy.
8.3. CRITICAL PROCESS PARAMETERS (CPPS):
Critical Process Parameters are manufacturing variables that significantly influence one or more Critical Quality Attributes and therefore require appropriate control during formulation development. In canagliflozin solid dispersion systems, processing conditions directly affect molecular dispersion of the drug, degree of amorphization, dissolution behaviour, and physical stability. The most important CPPs include drug-to-polymer ratio, solvent selection, mixing speed, mixing time, drying temperature, solvent evaporation rate, milling conditions, and compression force during tablet preparation. Variations in these parameters may alter particle morphology, residual solvent content, crystallinity, and dissolution performance. Therefore, optimization of processing conditions is essential to produce reproducible formulations with consistent pharmaceutical quality.
Within the QbD framework, CPPs are systematically investigated using statistical Design of Experiments, allowing the identification of optimal operating conditions while minimizing manufacturing variability.
8.4. FACTORIAL DESIGN MODELS:
Factorial experimental design is one of the most widely applied statistical optimization tools in pharmaceutical formulation development. It enables simultaneous evaluation of multiple formulation variables together with their interaction effects using a limited number of experimental runs.
8.4.1. 2² FACTORIAL DESIGN:
The 2² factorial design evaluates two independent formulation variables, each studied at two levels (low and high), resulting in four experimental formulations. In canagliflozin solid dispersion development, independent variables may include the concentration of PVP K30 and PEG 6000, whereas dependent responses include saturation solubility, percentage drug release, and dissolution efficiency. This experimental design identifies the individual contribution of each polymer as well as their interaction on formulation performance. Because relatively few experiments are required, the 2² factorial design is considered highly suitable for preliminary optimization studies and screening of formulation variables.
8.4.2. 3² Factorial Design
The 3² factorial design evaluates two formulation variables at three levels (low, medium, and high), generating nine experimental formulations. Compared with the 2² design, this approach provides a more detailed understanding of nonlinear relationships between formulation variables and pharmaceutical responses. In canagliflozin solid dispersion optimization, the concentration of hydrophilic carriers and drug-polymer ratio are frequently selected as independent variables, while dissolution rate, solubility enhancement, and drug content serve as response variables. Statistical analysis identifies the optimum polymer concentration required to maximize dissolution without compromising formulation stability.
8.5.Response Surface Methodology (RSM)
Response Surface Methodology is an advanced statistical optimization technique employed following factorial experimentation to develop mathematical models describing the relationship between formulation variables and pharmaceutical responses. The technique generates polynomial equations together with three-dimensional response surface and contour plots that illustrate the influence of independent variables on formulation performance. For canagliflozin solid dispersions, Response Surface Methodology facilitates simultaneous optimization of polymer concentration, carrier ratio, and processing conditions to maximize aqueous solubility, dissolution rate, and physical stability. The generated models predict formulation performance within the experimental design space, thereby reducing the number of confirmatory experiments required during product development. Consequently, RSM has become an indispensable component of Quality by Design because it provides scientific understanding, process optimization, and robust formulation development with improved regulatory acceptance.
9. FUTURE PERSPECTIVES AND RESEARCH OPPORTUNITIES:
The development of solid dispersion technology has significantly improved the solubility, dissolution rate, and oral bioavailability of poorly water-soluble drugs such as canagliflozin. Despite these advancements, several challenges remain, including long-term physical stability, scale-up, reproducibility, and prediction of optimal formulation variables. Emerging technologies such as artificial intelligence (AI), machine learning (ML), advanced polymeric carriers, and personalized medicine are expected to transform pharmaceutical formulation development by enabling more efficient, data-driven, and patient-centric approaches. These innovations offer new opportunities to improve formulation performance while reducing development time and manufacturing costs.
Future investigations should focus on the development of novel multifunctional polymeric carriers capable of simultaneously enhancing solubility, maintaining supersaturation, and preventing recrystallization during long-term storage. Advanced polymers such as Soluplus®, Copovidone, hydroxypropyl methylcellulose acetate succinate (HPMCAS), poloxamers, and amphiphilic graft copolymers possess superior stabilization properties and may further improve the dissolution behaviour and physical stability of canagliflozin solid dispersions. Hybrid carrier systems combining hydrophilic polymers with surfactants or lipid-based excipients may provide synergistic improvements in drug release and oral bioavailability.
Further research should also investigate alternative manufacturing technologies, including hot-melt extrusion, spray drying, freeze drying, electrospinning, and supercritical fluid processing. These advanced techniques offer improved control over particle morphology, molecular dispersion, and process reproducibility while facilitating industrial-scale production. Comparative evaluation of these preparation methods may identify the most suitable manufacturing approach for commercial development of canagliflozin solid dispersions.
CONCLUSION
Canagliflozin is an effective sodium-glucose co-transporter 2 (SGLT2) inhibitor widely used in the management of type 2 diabetes mellitus. However, its therapeutic performance is significantly limited by poor aqueous solubility, slow dissolution rate, and low oral bioavailability, characteristics associated with its Biopharmaceutics Classification System (BCS) Class IV classification. These physicochemical limitations necessitate the application of advanced formulation strategies capable of enhancing drug solubility, dissolution behaviour, and systemic absorption. This review comprehensively highlights the pharmaceutical challenges associated with canagliflozin and critically discusses the various formulation approaches investigated to overcome these limitations. Among the available solubility enhancement techniques, solid dispersion technology has emerged as one of the most effective and versatile approaches for improving the biopharmaceutical performance of canagliflozin. The incorporation of hydrophilic carriers such as polyvinylpyrrolidone (PVP K30), polyethylene glycol (PEG 6000), hydroxypropyl methylcellulose (HPMC), Poloxamers, Soluplus®, and Copovidone significantly enhances drug dissolution through multiple complementary mechanisms, including amorphization, improved wettability, particle size reduction, increased porosity, and inhibition of recrystallization. These physicochemical modifications facilitate rapid drug dissolution and improved gastrointestinal absorption, ultimately resulting in enhanced oral bioavailability and therapeutic efficacy.
The review further emphasizes the importance of comprehensive physicochemical characterization using analytical techniques such as Fourier Transform Infrared Spectroscopy (FTIR), Differential Scanning Calorimetry (DSC), Powder X-ray Diffraction (PXRD), and Scanning Electron Microscopy (SEM). These techniques provide valuable information regarding drug–polymer compatibility, crystallinity, thermal behaviour, surface morphology, and physical stability, thereby ensuring successful formulation development. Furthermore, the implementation of Quality by Design (QbD) principles and statistical optimization tools, including factorial design and response surface methodology, enables systematic optimization of formulation variables, enhances process understanding, and supports the development of robust and reproducible pharmaceutical products.
Despite substantial advancements, challenges related to long-term physical stability, large-scale manufacturing, and in vitro–in vivo correlation remain important areas for future investigation. Emerging technologies, including advanced multifunctional polymeric carriers, computational modelling, artificial intelligence, machine learning, and personalized pharmaceutical formulations, are expected to further accelerate the development of highly efficient solid dispersion systems. Overall, solid dispersion technology represents a scientifically robust and commercially promising strategy for overcoming the solubility limitations of canagliflozin. Continued research integrating innovative carrier systems, advanced manufacturing technologies, and predictive formulation approaches will facilitate the development of stable, high-performance oral dosage forms, ultimately improving therapeutic outcomes and patient care in the management of type 2 diabetes mellitus.
REFERENCES

Aniket Pawar, Parag Kothawde, Dr. Prashant Malpure, Rohan Tarle, Sanket Mogal, Solid Dispersion-Based Strategies for Solubility Enhancement of Canagliflozin: A Comprehensive Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 32-60, https://doi.org/10.5281/zenodo.21103141
 10.5281/zenodo.21103141
10.5281/zenodo.21103141