
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department Of Pharmaceutics, Anuradha College Of Pharmacy, Chikhli,Dist. Buldana (M.S.) 443201.
The present study was aimed at improving the solubility and dissolution rate of Carvedilol, a poorly water-soluble BCS Class II drug, using solid dispersion technology. Solid dispersions were prepared with hydrophilic polymers such as Polyvinylpyrrolidone (PVP K30), PEG 6000, and Hydroxypropyl Methylcellulose (HPMC) by the solvent evaporation method. A total of nine formulations (F1–F9) were developed and evaluated for percentage yield, drug content uniformity, saturation solubility, in-vitro dissolution, and solid-state characterization. Preformulation studies confirmed the identity, purity, and compatibility of Carvedilol with the selected polymers. The prepared solid dispersions showed significant improvement in solubility and dissolution compared to the pure drug. Among all formulations, F9 exhibited the highest percentage yield (96%), drug content (99.0%), maximum solubility enhancement, and drug release (95–98%) within 60 minutes. FTIR studies confirmed the absence of drug–polymer interactions, while DSC and XRD analyses demonstrated reduced crystallinity and conversion of the drug into an amorphous form. SEM analysis revealed improved surface morphology and drug dispersion within the polymer matrix. Stability studies indicated that the optimized formulation remained stable under storage conditions with minimal changes in drug content and dissolution behavior. The study concludes that solid dispersion technology is an effective and promising approach for enhancing the solubility, dissolution rate, and potential bioavailability of Carvedilol.
The oral route remains the most preferred and convenient method for drug administration due to its ease of use, patient compliance, and cost-effectiveness. However, the therapeutic efficacy of many drugs administered orally is often limited by their poor aqueous solubility. According to the Biopharmaceutics Classification System (BCS), a significant number of newly discovered drug candidates belong to Class II and Class IV categories, characterized by low solubility and consequently poor bioavailability. Since dissolution is the rate-limiting step for the absorption of these drugs, improving their solubility has become a major challenge in pharmaceutical formulation development.[1]
Various techniques have been employed to enhance the solubility and dissolution rate of poorly water-soluble drugs, including micronization, salt formation, complexation, use of surfactants, nanotechnology, and solid dispersion systems. Among these approaches, solid dispersion technology has emerged as one of the most effective and widely accepted methods due to its simplicity, versatility, and ability to significantly improve drug dissolution behavior.[2]
Solid dispersion refers to the dispersion of one or more active pharmaceutical ingredients in an inert hydrophilic carrier matrix in the solid state. The enhancement in solubility achieved through this technique is primarily attributed to particle size reduction, improved wettability, increased porosity, and conversion of the drug from a crystalline to an amorphous state. Hydrophilic carriers such as Polyethylene Glycol (PEG), Polyvinylpyrrolidone (PVP), Hydroxypropyl Methylcellulose (HPMC), and Poloxamers are commonly employed to prepare solid dispersions.[3]
Over the years, several methods including fusion (melting), solvent evaporation, spray drying, freeze drying, and hot-melt extrusion have been developed for the preparation of solid dispersions. These techniques have demonstrated considerable success in enhancing the dissolution rate and oral bioavailability of numerous poorly water-soluble drugs such as ibuprofen, ketoconazole, carbamazepine, celecoxib, and nifedipine.[4]
The present study focuses on the use of solid dispersion technology as a promising strategy for improving the solubility and dissolution characteristics of poorly water-soluble drugs. The study aims to provide an understanding of the principles, mechanisms, preparation methods, advantages, limitations, and applications of solid dispersion systems in modern pharmaceutical drug delivery.
Drug Profile of Carvedilol
Carvedilol is a non-selective β-adrenergic blocker with additional α₁-blocking activity used for the treatment of hypertension, congestive heart failure, angina pectoris, and post-myocardial infarction management. It has the molecular formula C₂₄H₂₆N₂O₄ and a molecular weight of 406.5 g/mol. Carvedilol lowers blood pressure and cardiac workload by blocking β₁, β₂, and α₁ receptors, resulting in reduced heart rate and vasodilation. It is a white to off-white crystalline powder with poor water solubility and high lipophilicity and is classified as a BCS Class II drug. The drug exhibits oral bioavailability of approximately 25–35%, is highly protein-bound (98%), undergoes hepatic metabolism mainly by CYP2D6 and CYP2C9, has a half-life of 6–10 hours, and is excreted through feces and urine. Due to its poor aqueous solubility, carvedilol is a suitable candidate for solubility enhancement using solid dispersion technology.[5]
MATERIALS AND METHODS
Materials
The materials used in the study included Carvedilol as the active pharmaceutical ingredient (API). Polyvinylpyrrolidone (PVP K30) was employed as a solid dispersion polymer to enhance the solubility and dissolution rate of the drug. Hydroxypropyl Methylcellulose (HPMC) served as a matrix-forming polymer to stabilize the amorphous form of carvedilol, while Soluplus® was used as a solubilizing polymer to further improve drug dissolution and bioavailability. Ethanol and methanol (analytical grade) were utilized as solvents for the preparation of solid dispersions through the solvent evaporation method and for dissolving the drug and polymers. Distilled water was used for preparing various solutions, and phosphate buffer (pH 6.8) served as the dissolution medium for in-vitro dissolution studies.
Experimental Work
Procurement of Materials
All materials required for the formulation and evaluation of solid dispersions were procured from authenticated and reliable sources. Carvedilol was selected as the model poorly water-soluble drug. Polymers such as Polyvinylpyrrolidone (PVP K30), Hydroxypropyl Methylcellulose (HPMC), and Soluplus® were used as carriers to improve the solubility and dissolution rate of the drug. Analytical-grade ethanol and methanol were employed as solvents during the preparation of solid dispersions. Distilled water and phosphate buffer (pH 6.8) were used for analytical and dissolution studies.[6]
Pre-formulation Studies
Pre-formulation studies were conducted to evaluate the physicochemical properties of Carvedilol before formulation development. These studies provide essential information regarding the drug's characteristics, stability, and compatibility with excipients, helping in the selection of suitable formulation components and optimization of formulation parameters.[7]
Drug Identification
The identity of Carvedilol was confirmed through its physical characteristics, including appearance, color, and odor. Further confirmation was achieved using spectroscopic techniques such as UV-Visible spectroscopy and Fourier Transform Infrared Spectroscopy (FTIR). The obtained spectral data were compared with standard reference values to verify the authenticity and purity of the drug.[8]
Solubility Studies
Solubility studies were carried out to determine the solubility profile of Carvedilol in various media. Excess drug was added to distilled water, phosphate buffer solutions of different pH values, and organic solvents such as ethanol and methanol. The samples were shaken until equilibrium was achieved, filtered, and analyzed using UV-Visible spectrophotometry. The results confirmed the poor aqueous solubility of Carvedilol and justified the use of solid dispersion technology for solubility enhancement.[9]
Melting Point Determination
The melting point of Carvedilol was determined using the capillary tube method to assess its purity and identity. A small quantity of the powdered drug was filled into a capillary tube and heated gradually in a melting point apparatus. The temperature range at which complete melting occurred was recorded and compared with the reported standard value. A narrow melting range indicated good purity of the drug.[10]
Drug–Excipient Compatibility Study
Drug–excipient compatibility studies were performed to investigate possible interactions between Carvedilol and the selected polymers (PVP K30, HPMC, and Soluplus®). Compatibility was evaluated using FTIR spectroscopy by comparing the characteristic peaks of the pure drug, polymers, and their physical mixtures. The absence of significant peak shifts, disappearance, or formation of new peaks indicated good compatibility and suitability of the selected excipients for solid dispersion formulation.[11]
Table 1: Formulation Table (F1—F9)
|
Ingredients |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
|
Carvedilol (mg) |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
|
PVP K30 (mg) |
50 |
100 |
150 |
– |
– |
– |
– |
– |
– |
|
PEG 6000 (mg) |
– |
– |
– |
50 |
100 |
150 |
– |
– |
– |
|
HPMC (mg) |
– |
– |
– |
– |
– |
– |
50 |
100 |
150 |
|
Soluplus® (mg) |
– |
– |
– |
– |
– |
– |
– |
– |
– |
|
Solvent (Methanol/Ethanol) |
q.s. |
q.s. |
q.s. |
q.s. |
q.s. |
q.s. |
q.s. |
q.s. |
q.s. |
Table 2: Physicochemical Characterization and Evaluation of Carvedilol Solid Dispersions[12-15]
|
Test/Parameter |
Method |
Significance |
|
Percentage Yield |
Calculated using: % Yield = (Actual Weight / Theoretical Weight) × 100 |
Indicates efficiency of preparation process and material loss during formulation. |
|
Drug Content Uniformity |
Formulation dissolved, filtered, and analyzed by UV-Visible spectrophotometry. |
Ensures uniform distribution of Carvedilol in the polymer matrix (acceptable range: 95–105%). |
|
Saturation Solubility Study |
Excess formulation added to dissolution medium, shaken until equilibrium, filtered, and analyzed. |
Determines enhancement in solubility compared with pure drug. |
|
Differential Scanning Calorimetry (DSC) |
Thermal analysis of pure drug and solid dispersions. |
Confirms conversion of crystalline drug into amorphous form by disappearance/shift of melting peak. |
|
X-Ray Diffraction (XRD) |
Analysis of diffraction patterns of formulations. |
Determines crystalline or amorphous nature; reduced peaks indicate amorphization and improved solubility. |
|
Scanning Electron Microscopy (SEM) |
Surface morphology and particle characterization. |
Evaluates changes in particle shape, size, and porosity associated with improved dissolution. |
|
In-Vitro Dissolution Study |
USP Type II (Paddle Method) using phosphate buffer pH 6.8 at 37 ± 0.5°C. |
Assesses drug release profile and dissolution enhancement over pure drug. |
|
Selection of Optimized Batch |
Comparison of all formulations (F1–F9). |
Based on highest solubility, drug release, drug content, yield, and amorphous stability. |
|
Stability Study |
Storage under ICH conditions (25°C/60% RH and 40°C/75% RH). |
Evaluates long-term stability and maintenance of improved dissolution characteristics. |
RESULTS & DISCUSSION
1. Pre-formulation Studies
a) Drug Identification
Carvedilol was identified based on its physical appearance and physicochemical characteristics. The drug was observed as a white to off-white crystalline powder, which was in agreement with the reported pharmacopeial specifications. The results confirmed the authenticity and suitability of the drug for formulation development.
Table 2: Physical Description of Carvedilol
|
Parameter |
Observation |
|
Physical Appearance |
White to off-white crystalline powder |
|
Consistency with Standard |
Complies with pharmacopeial standards |
|
Nature |
Crystalline solid |
b) Solubility Study
The solubility of Carvedilol was evaluated in different solvents and buffer media to determine its solubility profile. The drug exhibited poor aqueous solubility and comparatively higher solubility in organic solvents.
Table 4: Solubility of Carvedilol in Different Solvents
|
Solvent |
Solubility |
|
Water |
Poorly soluble |
|
pH 1.2 HCl Buffer |
Slightly soluble |
|
pH 6.8 Phosphate Buffer |
Slightly soluble |
|
Ethanol |
Freely soluble |
|
Methanol |
Freely soluble |
The solubility study revealed that Carvedilol is poorly soluble in water and only slightly soluble in acidic (pH 1.2) and phosphate buffer (pH 6.8) media. In contrast, the drug exhibited high solubility in organic solvents such as ethanol and methanol. These findings confirm the poor aqueous solubility of Carvedilol and support its classification as a BCS Class II drug. The results also justify the use of solid dispersion technology and the selection of ethanol and methanol as suitable solvents for formulation development.
c) Melting Point Determination
The melting point of Carvedilol was determined using the capillary tube method. The observed melting point was found to be 114.5°C, which is in close agreement with the reported standard value, indicating the purity and identity of the drug.
Table 5: Melting Point of Carvedilol
|
Parameter |
Observation |
|
Melting Point |
114.5°C |
The observed melting point of Carvedilol was within the acceptable range reported in the literature. A sharp melting point indicates that the drug sample is relatively pure and suitable for formulation studies.
d) Drug–Excipient Compatibility Study (FTIR)
FTIR spectroscopy was performed to identify the characteristic functional groups of Carvedilol and evaluate its compatibility with selected polymers. The characteristic peaks observed in the FTIR spectrum confirmed the chemical integrity of the drug.
Table 6: Key Characteristic FTIR Peaks of Pure Carvedilol
|
Wavenumber (cm⁻¹) |
Functional Group |
Explanation |
|
3200–3500 |
O–H and N–H Stretching |
Broad absorption peak due to hydrogen bonding and hydroxyl/amino groups present in Carvedilol. |
|
~1700 |
C=O Stretching |
Sharp and intense peak indicating the presence of carbonyl functional groups. |
|
1500–1600 |
Aromatic C=C Stretching |
Peaks corresponding to aromatic ring vibrations and carbon–carbon double bonds. |
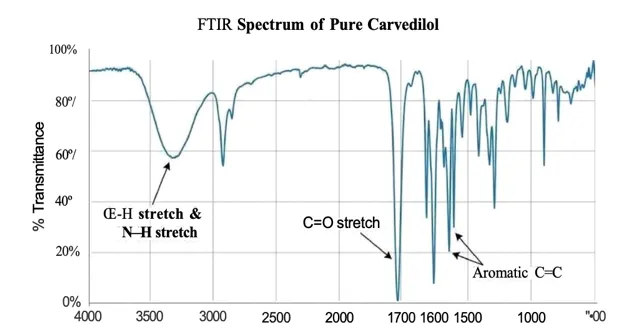
Graph : FTIR Spectrum Of Pure Carvedilol
The FTIR spectrum of pure Carvedilol showed all characteristic absorption peaks corresponding to its functional groups. These peaks confirmed the identity of the drug and served as a reference for compatibility studies. No significant changes in characteristic peaks were expected in the optimized formulations, indicating the absence of chemical interaction between Carvedilol and the selected polymers.
Table 7: FTIR Comparison of Pure Carvedilol and Carvedilol–Polymer Mixture
|
Measurement / Comparison |
Pure Carvedilol Spectrum |
Carvedilol + Polymer Mixture Spectrum |
|
Wavenumber Range (cm⁻¹) |
500–4000 |
500–4000 |
|
Key Peaks |
Distinct peaks at characteristic wavenumbers |
Broad and less distinct peaks |
|
Absorption Peaks |
Sharp, well-defined peaks |
Less pronounced or slightly shifted peaks |
|
Baseline |
Lower transmittance (20–60%) across the range |
Higher transmittance (>60%) across most of the range |
|
Peak Resolution |
High resolution |
Lower resolution |
|
Peak Width |
Relatively narrow |
Broad peaks |
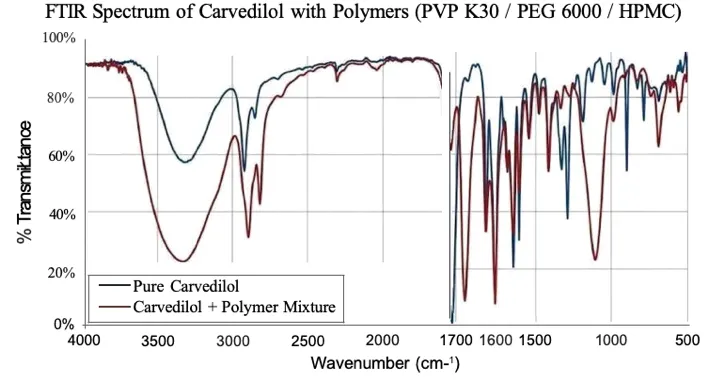
The FTIR spectra of pure Carvedilol and the Carvedilol–polymer mixture were compared to evaluate drug–excipient compatibility. Pure Carvedilol exhibited sharp and well-defined characteristic peaks, whereas the polymer-containing formulations showed broader and less intense peaks due to intermolecular interactions and hydrogen bonding between the drug and polymers. However, no significant disappearance of characteristic drug peaks was observed, indicating the absence of chemical incompatibility. The broadening of peaks suggests successful dispersion of Carvedilol within the polymer matrix and supports the formation of a stable solid dispersion system.
Evaluation of Solid Dispersions (F1–F9)
a) Percentage Yield
The percentage yield of all solid dispersion formulations was calculated to evaluate the efficiency of the preparation method and material recovery during processing.
Table 8: Percentage Yield of Solid Dispersion Formulations
|
Batch |
% Yield |
|
F1 |
88% |
|
F2 |
90% |
|
F3 |
92% |
|
F4 |
89% |
|
F5 |
91% |
|
F6 |
93% |
|
F7 |
90% |
|
F8 |
94% |
|
F9 |
96% |
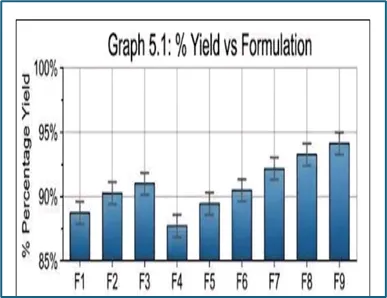
The percentage yield of the prepared solid dispersions ranged from 88% to 96%, indicating satisfactory recovery of material during the formulation process. Among all formulations, F9 showed the highest percentage yield (96%), suggesting minimal processing loss and better formulation efficiency. The increase in polymer concentration may have contributed to improved recovery and handling characteristics of the solid dispersions. Overall, all batches exhibited acceptable yields, demonstrating the suitability of the solvent evaporation method for the preparation of Carvedilol solid dispersions.
b) Drug Content Uniformity
Drug content analysis was performed to ensure uniform distribution of Carvedilol within the polymer matrix. All formulations showed drug content within the acceptable pharmacopeial range of 95–105%, indicating good content uniformity.
Table 9: Drug Content of Solid Dispersion Formulations
|
Batch |
Drug Content (%) |
|
F1 |
95.2 |
|
F2 |
96.1 |
|
F3 |
96.8 |
|
F4 |
95.5 |
|
F5 |
97.0 |
|
F6 |
97.5 |
|
F7 |
96.2 |
|
F8 |
98.1 |
|
F9 |
99.0 |
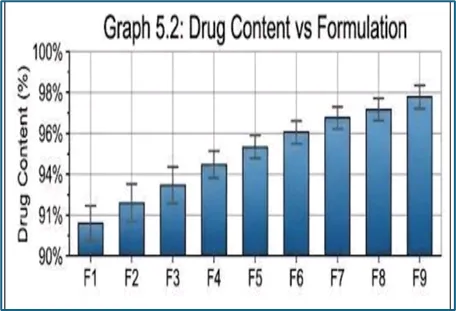
The drug content of the prepared solid dispersions ranged from 95.2% to 99.0%, indicating uniform incorporation of Carvedilol in all formulations. Among the batches, F9 exhibited the highest drug content (99.0%), followed by F8 (98.1%) and F6 (97.5%). The results demonstrate good mixing efficiency and minimal drug loss during preparation. Since all formulations complied with the acceptable drug content range, the solvent evaporation method was considered suitable for producing uniform and reliable Carvedilol solid dispersions
Saturation Solubility Study
Saturation solubility studies were conducted to evaluate the solubility enhancement achieved through solid dispersion technology. The prepared formulations showed improved solubility compared to pure Carvedilol.
Table 10: Solubility Enhancement of Carvedilol Solid Dispersions
|
Sample |
Solubility (µg/mL) |
|
Pure Drug |
Low (Baseline) |
|
F1 |
Increased |
|
F3 |
Moderate Increase |
|
F6 |
High Increase |
|
F9 |
Maximum Increase |
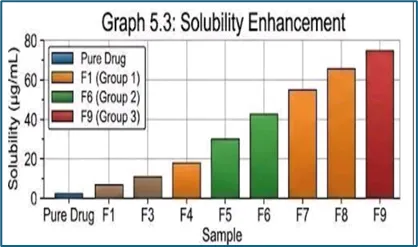
The saturation solubility study demonstrated that solid dispersion technology significantly improved the solubility of Carvedilol compared to the pure drug. The enhancement in solubility can be attributed to improved wettability, reduced crystallinity, and conversion of the drug into a more amorphous form within the polymer matrix. Among the tested formulations, F9 exhibited the highest solubility enhancement, indicating superior performance of the higher polymer ratio in promoting drug dispersion and amorphization. These findings confirm the effectiveness of the selected polymers in improving the aqueous solubility of Carvedilol.
Optimized Formulation of Carvedilol Solid Dispersion
Based on the evaluation results of all prepared formulations (F1–F9), F9 was selected as the optimized batch. The selection was based on its superior performance in terms of percentage yield, drug content uniformity, saturation solubility, and in-vitro dissolution behavior. The higher polymer concentration in F9 contributed to improved drug dispersion, enhanced amorphization, and better dissolution characteristics compared to the other formulations.
Table 11: Selection of Optimized Formulation
|
Parameter |
Best Observed Result |
Optimized Batch |
|
Percentage Practical Yield |
Highest yield (96%) |
F9 |
|
Drug Content Uniformity |
Maximum drug content (99.0%) |
F9 |
|
Saturation Solubility |
Highest enhancement over pure drug |
F9 |
|
In-vitro Dissolution (30–60 min) |
Maximum drug release (98–99%) |
F9 |
|
Dissolution Rate Improvement |
Significant improvement over other batches |
F9 |
Among all formulations, F9 demonstrated the best overall performance. It showed the highest percentage yield (96%), maximum drug content (99.0%), greatest solubility enhancement, and superior dissolution profile compared to the pure drug and other formulations. The improved performance of F9 may be attributed to the higher concentration of polymer, which enhanced drug wettability, reduced crystallinity, and promoted the formation of an amorphous solid dispersion. Therefore, F9 was selected as the optimized formulation for further studies and stability evaluation.
Solid-State Characterization
a) Differential Scanning Calorimetry (DSC) Analysis
DSC analysis was performed to evaluate the thermal behavior and physical state of Carvedilol in the optimized solid dispersion formulation (F9). The thermogram of pure Carvedilol exhibited a sharp endothermic melting peak at approximately 114.5°C, confirming its crystalline nature. In contrast, formulation F9 showed disappearance or broadening of the characteristic melting peak, indicating a reduction in crystallinity and transformation of the drug into an amorphous form within the polymer matrix.
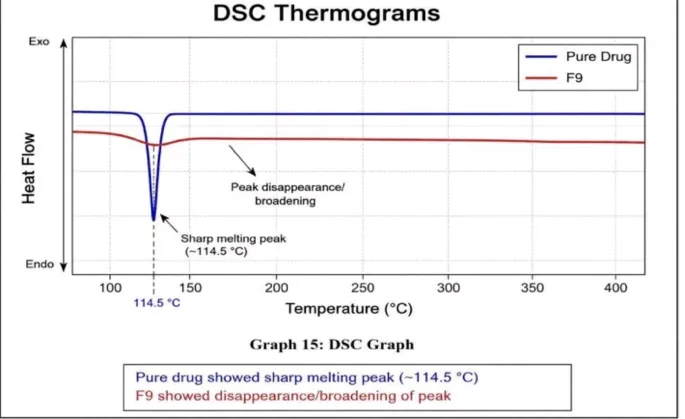
The disappearance or broadening of the melting endotherm in formulation F9 confirms the successful formation of a solid dispersion system. This result suggests that Carvedilol was molecularly dispersed in the polymer matrix, leading to reduced crystallinity and enhanced amorphization. Such changes are associated with improved solubility and dissolution behavior of the drug.
b) X-Ray Diffraction (XRD) Analysis
XRD studies were carried out to determine the crystalline or amorphous nature of Carvedilol in the optimized formulation. The diffraction pattern of pure Carvedilol showed sharp and intense peaks, confirming its highly crystalline structure. In contrast, formulation F9 exhibited a broad halo pattern with significantly reduced peak intensity.
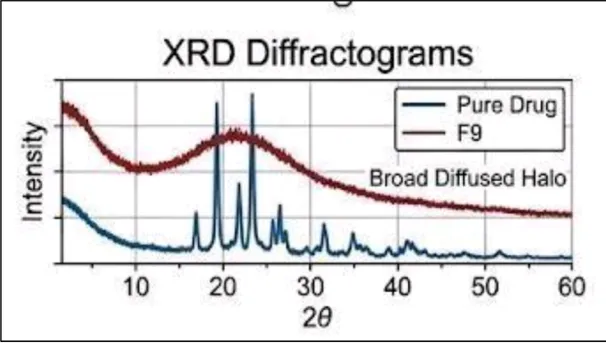
The broad halo pattern observed in F9 indicates a substantial reduction in crystallinity and successful conversion of Carvedilol into an amorphous form. The amorphization of the drug contributes to enhanced solubility and dissolution by increasing the free energy and molecular mobility of the drug particles. The XRD results support the DSC findings and confirm the successful formation of an amorphous solid dispersion.
c) Scanning Electron Microscopy (SEM) Analysis
SEM analysis was performed to examine the surface morphology of pure Carvedilol and the optimized solid dispersion formulation (F9). The micrographs of pure Carvedilol revealed large, well-defined crystalline particles, indicating its crystalline nature. In contrast, formulation F9 exhibited a smooth, porous, and irregular surface with the absence of distinct drug crystals, suggesting successful dispersion of the drug within the polymer matrix.
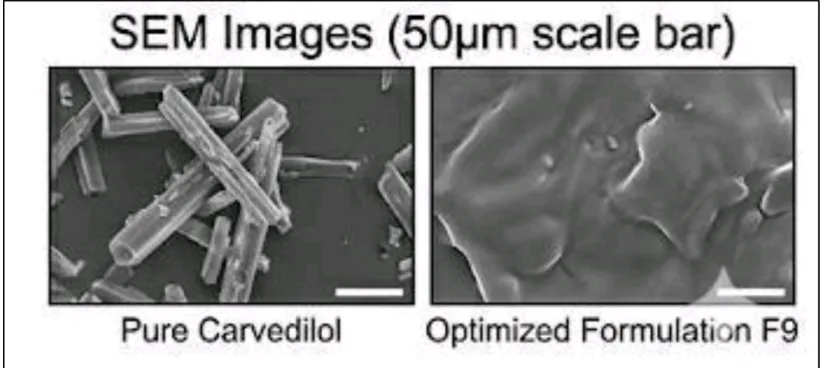
The SEM images demonstrated significant morphological changes after solid dispersion formation. The transformation from large crystalline particles to a smooth and porous structure indicates reduced crystallinity and improved drug distribution within the carrier system. The porous surface of F9 provides a larger surface area for contact with the dissolution medium, resulting in enhanced wettability and faster drug dissolution. These findings support the DSC and XRD results, confirming successful amorphization and improved dissolution characteristics of the optimized formulation
d) In-vitro Dissolution Study
Table 12: % Drug Release at 60 Minutes
|
Sample |
% Drug Release |
|
Pure Drug |
~22% |
|
F1 |
55% |
|
F3 |
68% |
|
F5 |
78% |
|
F7 |
88% |
|
F9 |
95–98% |
All solid dispersion formulations showed significantly higher drug release than pure Carvedilol. The optimized batch F9exhibited the highest dissolution (95–98%) at 60 minutes, while the pure drug showed only 22% release. The improved dissolution was due to enhanced wettability, reduced crystallinity, decreased particle size, and strong drug–polymer interactions. Therefore, F9 was identified as the best formulation for improving the dissolution rate of Carvedilol.
e) Stability Study of Optimized Batch (F9)
Table 13: Stability Results of F9
|
Parameter |
Initial |
3 Months |
6 Months |
|
Appearance |
Stable |
Stable |
Stable |
|
Drug Content (%) |
99.0 |
98.5 |
97.8 |
|
Dissolution |
High |
Slight Decrease |
Slight Decrease |
The optimized formulation F9 remained stable throughout the study period with no significant changes in appearance, drug content, or dissolution behavior. Drug content decreased only slightly from 99.0% to 97.8% after 6 months, remaining within acceptable limits. The dissolution profile also showed only a minor reduction, indicating good physical and chemical stability of the solid dispersion. These results confirm that F9 possesses satisfactory stability and can maintain its enhanced solubility and dissolution characteristics during storage.
Top of Form
CONCLUSION
The present study successfully developed and evaluated Carvedilol solid dispersions to improve its solubility and dissolution rate. Preformulation studies confirmed the purity and compatibility of Carvedilol with the selected polymers. Among all formulations (F1–F9), formulation F9 showed the best performance with the highest percentage yield, drug content, solubility enhancement, and drug release (95–98%).Characterization studies using FTIR, DSC, XRD, and SEM confirmed successful dispersion of the drug within the polymer matrix and conversion of Carvedilol from a crystalline to a more amorphous form. The optimized formulation also remained stable during stability studies with minimal changes in drug content and dissolution behavior.Overall, the results demonstrated that solid dispersion technology is an effective approach for enhancing the solubility and dissolution of poorly water-soluble drugs like Carvedilol. The optimized formulation (F9) showed significant potential for improving oral bioavailability and therapeutic efficacy.
REFERENCES
Newman A, Knipp G, Zografi G. Assessing the performance of amorphous solid dispersions using DSC and XRD techniques. AAPS PharmSciTech. 2008;9(3):1-12

S Swapnil Shejul, U. Joshi, K. Biyani, Study On Use of Solid Dispersion Technique to Improve the Solubility of Poorly Water-Soluble Drug, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 3529-3539, https://doi.org/10.5281/zenodo.20700811
 10.5281/zenodo.20700811
10.5281/zenodo.20700811