
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
S.N. College of Pharmacy Jaunpur UP.
Analytical Quality by Design (AQbD) is a systematic, science- and risk-based approach Vilanterol has been recognized as a gold standard, ultra-long-acting therapeutic agent in the management of chronic obstructive pulmonary disease (COPD) and asthma, and as a vital scaffold during advanced respiratory drug development. The current focus of the project was the rational design of a novel structural derivative utilizing vilanterol as the core substrate to systematically optimize its pharmacokinetic and receptor-binding profile. This study aimed to develop a novel compound incorporating a highly lipophilic N-methyl tertiary amine moiety with potential extended biological activity, assessing its mechanism as a robust long-acting ?2-adrenoceptor agonist.The synthesized moiety underwent comprehensive physicochemical evaluation to determine its percentage yield, melting point, and solubility. The target analogue yielded nearly 76 %, exhibited a melting point of 118-122 °C, and was freely soluble in DMSO and chloroform. The final target analogue was efficiently synthesized via a highly chemoselective reductive amination pathway. This involved the initial condensation of the vilanterol free base with aqueous formaldehyde, followed by direct reduction utilizing sodium triacetoxyborohydride under mildly acidic conditions.The sample was prepared as KBr pellets, and the FTIR spectrum of the derivative was recorded and analyzed. The FT-IR spectral characteristics of the analogue were specifically examined as 3385 cm?¹ (broad O-H Stretch), 2860-2935 cm?¹ (Aliphatic C-H Stretch), 2795 cm?¹ (N-CH3 Tertiary Amine Stretch), 1505-1610 cm?¹ (Aromatic C=C ring Stretch), 1115 cm?¹ (Ether C-O-C Stretch), and 765 cm?¹ (Aromatic C-Cl Stretch). NMR spectra were recorded at 400 MHz using DMSO-d6 as the solvent to confirm chemical shift values and elucidate structural information, showing characteristic shifts at ? 2.38 (singlet, 3H, N-CH3), ? 6.72-7.25 (Saligenin protons), and ? 7.40-7.55 (2,6-Dichlorophenyl protons). Additionally, ESI-MS (positive ion mode) was utilized to confirm the exact mass, revealing a pseudo-molecular ion base peak at m/z 500.2 [M+H]+, alongside a di-chlorinated M+2 isotopic peak at m/z 502.2 with high precision. “In this study, the resulting novel N-methyl vilanterol derivative was assessed for its successful synthesis and therapeutic potential. The synthesized molecule holds significant promise as a safe, effective, and ultra-long-acting respiratory bronchodilator, warranting further investigation into its specific pulmonary receptor kinetics and sustained anti-inflammatory pathways against obstructive airway diseases.”
1.1. The Global Burden of Obstructive Airway Diseases
Obstructive pulmonary conditions, like asthma and Chronic Obstructive Pulmonary Disease (COPD), are a huge and growing problem for public health around the world. These long-term lung diseases cause limited breathing, ongoing inflammation of the airways, and changes in the structure of the respiratory tract. COPD is the main cause of illness and death around the world. COPD is more common in countries that are industrialising quickly, like India. This is because of a lot of different risk factors, such as smoking tobacco, being exposed to biomass fuel smoke for a long time, and rising levels of particulate air pollution in cities [1].
Asthma, typically characterized by reversible bronchoconstriction and airway hyper-responsiveness, and COPD, defined by progressive and largely irreversible airflow limitation, both share a common therapeutic requirement: the need for effective, long-lasting bronchodilation. The architectural changes in the lungs, such as goblet cell hyperplasia, smooth muscle hypertrophy, and alveolar destruction-necessitate pharmacological interventions that can consistently maintain airway patency, reduce the frequency of severe exacerbations, and improve the patient's overall quality of life [2]. Consequently, the cornerstone of symptomatic management for these respiratory pathologies relies heavily on inhaled bronchodilators, with β2-adrenoceptor agonists serving as the primary therapeutic class.
1.2. Evolution and Pharmacology of β2-Adrenoceptor Agonists
Over the past few decades, the way drugs are used to treat bronchospasm has changed a lot. The β2-adrenoceptors are GPCRs, which are G-protein coupled receptors. They are mostly found on the smooth muscle cells in the airways. A substance called an agonist binds to the β2-adrenoceptor and causes a structural shift. This shift makes the stimulatory G-protein (Gs) alpha subunit exchange guanosine diphosphate (GDP) for guanosine triphosphate (GTP). This action speeds up the effector enzyme adenylate cyclase, which causes a rise in cyclic adenosine monophosphate (cAMP) inside cells. Protein Kinase A (PKA) is then activated by high cAMP, which phosphorylates a chain of target proteins. This eventually leads to the storage of calcium ions inside cells and the weakening of bronchial smooth muscle [4].
The historical progression of β2-agonists reflects a continuous refinement in medicinal chemistry aimed at maximizing receptor selectivity and prolonging the duration of action. Early non-selective agents, such as isoprenaline, provided effective bronchodilation but were severely limited by deleterious chronotropic and inotropic cardiovascular side effects due to concurrent β1-receptor activation in the myocardium. The development of Short-Acting β2-Agonists (SABAs) like salbutamol marked a breakthrough in selectivity, yet their short half-life necessitated frequent dosing. This limitation drove the design of Long-Acting β2-Agonists (LABAs) such as salmeterol and formoterol, providing 12-hour bronchodilation. Today, the clinical paradigm has shifted towards “Ultra-LABAs,” which offer a 24-hour duration of action, allowing for once-daily dosing regimens that dramatically improve patient compliance and therapeutic outcomes [4].
1.3. Structure-Activity Relationships (SAR) of Long-Acting Bronchodilators
The fundamental pharmacophore of β2-adrenoceptor agonists is derived from the endogenous catecholamine structure, consisting of a substituted aromatic ring, an ethanolamine backbone, and a basic secondary amine. The SAR of this class is exceptionally well-documented. The hydroxyl groups on the aromatic ring are critical for hydrogen bonding with serine residues (Ser203, Ser204, and Ser207) in transmembrane domain 5 (TM5) of the receptor. To circumvent rapid degradation by Catechol-O-Methyltransferase (COMT), the catechol moiety is often modified to a saligenin ring (as seen in salbutamol and vilanterol) or a resorcinol ring [5].
The chiral center at the β-carbon is stereoselective, with the R-enantiomer typically functioning as the eutomer, exhibiting vastly superior receptor affinity compared to the S-enantiomer. The most crucial determinant for both selectivity and duration of action lies in the substitution at the nitrogen atom. While bulky alkyl groups (like the tert-butyl group in salbutamol) confer β2 selectivity over β1 and provide resistance against degradation by Monoamine Oxidase (MAO), the attachment of long, highly lipophilic aralkyl chains drastically alters the pharmacokinetic profile. According to the “exosite” and “plasmalemma diffusion microkinetic” models, these extended lipophilic tails partition into the lipid bilayer of the cell membrane, serving as a drug reservoir, or bind to secondary, non-polar allosteric sites on the receptor, allowing the active headgroup to repeatedly engage and disengage the orthosteric site [3,4].
1.4. Vilanterol: Structural Profile and Clinical Significance
Vilanterol trifenatate is a highly potent, novel Ultra-LABA that exemplifies state-of-the-art bronchodilator design. It possesses a 24h duration of action and extraordinary selectivity for the β2-adrenoceptor. Structurally, vilanterol features a saligenin headgroup coupled to a highly optimized lipophilic tail: a hexamethylene linker attached to an ether oxygen, terminating in a 2,6-dichlorobenzyl group. This complex structural arrangement ensures that vilanterol rapidly partitions into the pulmonary tissue while maintaining a high intrinsic efficacy at the receptor site [2].
In clinical trials, vilanterol consistently improves forced expiratory volume in 1 second (FEV1) and greatly lowers the number of moderate to severe flare-ups in both COPD and asthma patients. This is especially true when it is combined in fixed doses with the inhaled corticosteroid fluticasone furoate. Furthermore, vilanterol was designed with an optimized systemic clearance profile; any fraction of the drug that is swallowed or absorbed into the systemic circulation is rapidly metabolized by hepatic cytochromes, minimizing the risk of systemic sympathomimetic side effects [2,3].
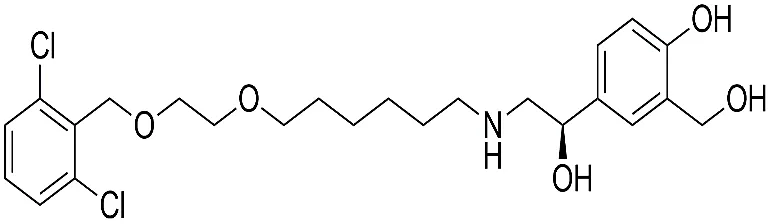
Figure 1.1: Chemical structure of Vilanterol.
1.5. Rationale for Drug Design: Exploring N-Methylation
While vilanterol represents a highly optimized molecule, the exploration of novel structural derivatives is a fundamental aspect of medicinal chemistry to continuously improve pharmacokinetic profiles, tissue residency, and receptor kinetics. The current thesis proposes the design and synthesis of an N-methyl derivative of vilanterol.
In classical β2-agonist SAR, the nitrogen atom is maintained as a secondary amine (NH). The protonated secondary amine at physiological pH is essential for forming a strong electrostatic ionic bond with the Aspartate 113 (Asp113) residue in TM3 of the receptor [5]. The conversion of this secondary amine to a tertiary amine via N-methylation represents a significant structural intervention. N-methylation subtly alters the basicity (pKa) of the nitrogen center while concurrently increasing the overall lipophilicity (log P) and steric bulk of the molecule.
This specific modification is hypothesized to influence the drug's spatial trajectory as it navigates from the lipid bilayer into the aqueous binding pocket. Furthermore, the incorporation of the N-methyl group may alter the metabolic stability of the compound, potentially slowing localized pulmonary degradation while preserving rapid systemic clearance. By synthesizing the N-methyl vilanterol derivative, this research aims to investigate whether a tertiary amine modification can maintain or enhance the sustained bronchodilatory effects afforded by the exosite-binding tail, whilst potentially modulating the onset of action and lipophilic tissue retention.
1.6. Synthetic Approaches to the N-Methyl Vilanterol Derivative
The synthetic strategy for the N-methyl vilanterol derivative will fundamentally rely on the selective alkylation of the existing secondary amine within the vilanterol scaffold, or a convergent synthesis building the tertiary amine from distinct precursor fragments.
The primary synthetic pathway involves the reductive amination of the vilanterol free base (or a suitably protected intermediate). Some weak reducing agents, like sodium cyanoborohydride (NaBH3CN) or sodium triacetoxyborohydride, should be present along with the secondary amine when this is done. The mixture should be slightly acidic to help the formation of iminium ions and the reduction that follows.
The successful execution of this synthesis will heavily depend on exact analytical characterization at every intermediate stage. The structural integrity of the synthesized N-methyl derivative will be verified using Fourier-Transform Infrared (FT-IR) spectroscopy to monitor the disappearance of the N-H stretching band and the emergence of relevant N-CH3 spectral signatures. High-resolution Proton Nuclear Magnetic Resonance (1H-NMR) spectroscopy will be critical for elucidating the proton environment, specifically isolating the singlet integration corresponding to the newly introduced methyl protons. Furthermore, Electrospray Ionization Mass Spectrometry (ESI-MS) will be deployed to definitively confirm the pseudo-molecular ion peaks (m/z), ensuring that precise mass assignments and elimination of background spectral noise are achieved to meet the rigorous reporting standards.
1.7. Aim and Objectives
Aim:
The primary aim of this research is to synthesize, and evaluate an N-methyl derivative of vilanterol as a novel, long-acting β2-adrenoceptor agonist.
Objectives:
LITERATURE SURVEY
Table 2.1: Drug Profile of Vilanterol.
|
S. No. |
Property |
Description |
|
1. |
Drug |
Vilanterol |
|
2. |
Molecular Formula |
C24H33Cl2NO5 |
|
3. |
Molecular Weight |
486.4 g/mol |
|
4. |
Elemental Composition |
C: 59.26%, H: 6.84%, Cl: 14.58%, N: 2.88%, O: 16.45% |
|
5. |
Preparation Method |
Multi-step synthesis involving the coupling of a protected (R)-saligenin epoxide (or related halohydrin) with an extended lipophilic amine chain terminating in a 2,6-dichlorobenzyl ether, followed by global deprotection. |
|
6. |
IUPAC Name |
4-[(1R)-2-[6-[2-[(2,6-dichlorophenyl)methoxy]ethoxy]hexylamino]-1-hydroxyethyl]-2-(hydroxymethyl)phenol |
|
7. |
Odor |
Odorless |
|
8. |
Colour |
White to off-white (solid powder) |
|
9. |
Solubility |
Practically insoluble in water; soluble in methanol, ethanol, and dimethyl sulfoxide (DMSO). |
|
10. |
Melting Point |
132°C - 135°C (Reported for Vilanterol trifenatate salt) |
|
11. |
Synonyms |
GW642444, N-[2-(4-hydroxy-3-hydroxymethylphenyl)-2-hydroxyethyl]-6-(2-(2,6-dichlorobenzyloxy)ethoxy)hexylamine |
|
12. |
pKa (at 25 °C) |
~ 9.7 (Predicted for the secondary amine); ~ 9.4 (Predicted for the phenolic -OH) |
|
13. |
Log P |
~ 4.1 (Calculated XLogP3, highly lipophilic) |
|
14. |
Structure |
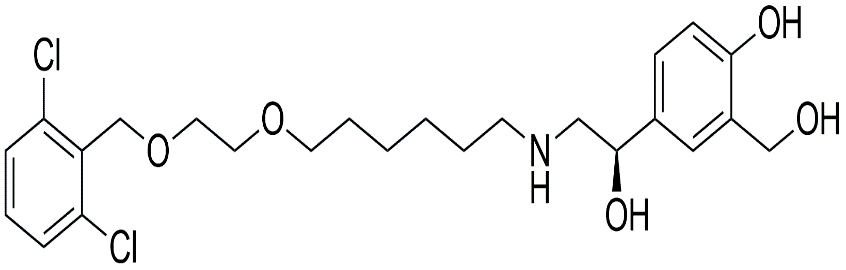
|
2.2. The Evolutionary Trajectory of Long-Acting Bronchodilators
Cazzola et al. (2013) provide a comprehensive overview of the evolutionary trajectory of bronchodilator therapy, tracking the transition from short-acting β2-agonists (SABAs) to long-acting (LABAs) and ultra-long-acting β2-agonists (Ultra-LABAs). The review highlights how the clinical demand for once-daily dosing regimens in asthma and COPD management drove the medicinal chemistry efforts to extend the duration of action beyond the 12h window of formoterol and salmeterol. The authors emphasize that achieving a 24h therapeutic duration fundamentally requires the integration of highly lipophilic side chains into the phenethanolamine scaffold. This structural modification facilitates prolonged retention within the pulmonary tissue or continuous re-engagement with the receptor. For the proposed thesis, this literature substantiates the foundational necessity of designing Ultra-LABA molecules that prioritize extended tissue residency and once-daily efficacy, thereby justifying the continued exploration and structural refinement of advanced scaffolds like vilanterol to optimize therapeutic outcomes and enhance patient compliance in chronic obstructive airway diseases. [4]
2.3. Rational Design and Discovery of the Vilanterol Scaffold
Procopiou et al. (2010) detail the pivotal medicinal chemistry campaign that culminated in the discovery of vilanterol. The paper delineates the rational design of a novel series of β2-adrenoceptor agonists utilizing an “antedrug” or “soft drug” approach. The researchers carefully looked at different lipophilic tails connected to a saligenin headgroup. They found that the 2,6-dichlorobenzyl ether moiety worked best for selectively targeting β2 and lasting for 24 hours. Importantly, the molecule was designed to be quickly broken down by liver cytochrome P450 enzymes, mainly by cutting the ether linker. This was done to reduce the typical cardiovascular side effects that are caused by the sympathetic nervous system. This research is instrumental for the thesis as it establishes the baseline structural parameters of the vilanterol scaffold. Understanding the delicate balance Procopiou and colleagues achieved between local pulmonary retention and rapid systemic clearance provides the critical comparative framework against which the pharmacokinetic alterations induced by the proposed N-methylation will be hypothesized, evaluated, and subsequently discussed. [3]
2.4. Pharmacokinetic Theories: Exosite and Microkinetic Models
Matera et al. (2007) critically examine the prevailing pharmacological theories explaining the extended duration of action inherent to long-acting and ultra-long-acting β2-adrenoceptor agonists. The paper juxtaposes two primary hypotheses: the “exosite theory,” where the lengthy lipophilic tail binds to a secondary, non-polar domain on the receptor preventing complete dissociation, and the “plasmalemma diffusion microkinetic model,” which suggests the drug partitions deeply into the cellular lipid bilayer, creating a localized depot that continuously supplies the active headgroup to the receptor’s orthosteric binding site. This literature is highly pertinent to the thesis as it provides the theoretical mechanism for how bulky, lipophilic modifications sustain bronchodilation. Modifying the vilanterol scaffold via N-methylation will inherently increase the molecule's overall lipophilicity (log P) and alter its three-dimensional spatial conformation. Consequently, this paper will serve as the theoretical foundation to hypothesize whether the addition of a tertiary amine disrupts or enhances these established microkinetic interactions within the pulmonary lipid milieu. [6]
2.5. Structural Significance of the Amine Core in Receptor Binding
Rasmussen et al. (2011) elucidated the high-resolution crystal structure of the human β2-adrenergic G-protein-coupled receptor in complex with an agonist, providing an unprecedented, three-dimensional understanding of the orthosteric binding pocket. The crystallographic data definitively demonstrates that the protonated amine of the β2-agonist forms a crucial ionic bridge and hydrogen-bonding network with the highly conserved Aspartate 113 (Asp113) residue located on transmembrane helix 3 (TM3). This specific intermolecular interaction is absolutely fundamental for anchoring the ligand within the receptor and initiating the conformational changes required for subsequent G-protein activation. This structural biology insight is exceptionally relevant to the proposed thesis. Because the project involves synthesizing an N-methyl derivative, thereby transforming a secondary amine into a tertiary amine, this paper provides the structural context to evaluate how increased steric bulk and altered basicity (pKa) at the nitrogen center might influence the binding affinity and the crucial electrostatic interaction with the key Asp113 residue. [7]
2.6. The Pharmacological Impact of N-Methylation in Drug Design
Schönherr and Cernak (2013) provide a profound analysis of the “magic methyl” effect and the specific impact of N-methylation in contemporary drug discovery and structural optimization. The review articulates how the conversion of a secondary amine to a tertiary amine via the introduction of a seemingly simple methyl group can drastically alter a drug’s physicochemical and pharmacokinetic properties. The authors highlight that N-methylation typically eliminates a hydrogen-bond donor, increases overall lipophilicity, alters the compound’s basicity, and can significantly restrict the conformational flexibility of the molecule. For the current thesis, this literature provides the core rational justification for the proposed synthetic modification of vilanterol. By applying Schönherr and Cernak’s principles, the literature review will argue that N-methylating the vilanterol scaffold is a strategic, calculated manipulation intended to modulate membrane permeability, optimize the spatial geometry of the lipophilic tail, and potentially alter the molecule’s metabolic vulnerability, thereby thoroughly justifying the targeted synthetic objectives of the research project. [8]
2.7. Clinical Efficacy and Safety Benchmarks of Vilanterol
Rayner and Scott (2014) comprehensively evaluate the clinical profile, efficacy, and safety of vilanterol, particularly when administered in combination with the inhaled corticosteroid fluticasone furoate for the management of COPD. The review thoroughly examines late-stage clinical trial data, affirming that vilanterol produces rapid bronchodilation comparable to salbutamol while reliably sustaining improvements in the forced expiratory volume in one second (FEV1) over a full 24h period. Furthermore, the paper highlights the compound’s excellent safety and tolerability profile, noting the minimal incidence of systemic sympathomimetic side effects such as tachycardia or tremors. This clinical literature is vital for the thesis as it establishes the exceptionally high benchmark of safety and efficacy that any novel vilanterol derivative must strive to achieve or exceed. It sets the clinical context, ensuring that the pharmacological evaluation of the synthesized N-methyl derivative is strictly compared against the established therapeutic parameters and systemic clearance expectations of the parent compound. [2]
2.8. Relevant Preclinical In Vivo Models for Assessing Airway Pharmacology
Glaab et al. (2007) provide a critical, comparative evaluation of invasive and non-invasive methodologies for assessing airway hyper-responsiveness and lung function in murine models of pulmonary disease. The paper details techniques such as whole-body plethysmography and invasive mechanical ventilation, emphasizing the precision required to accurately measure airway resistance and dynamic compliance in response to bronchoconstrictors like methacholine. The authors discuss the translational reliability of these murine models in evaluating the efficacy of novel bronchodilator therapeutics. This paper is highly relevant to the pharmacological evaluation chapter of the proposed thesis. [9]
2.9. Interrogating the Inflammatory Axis in Obstructive Airway Disease
Barnes (2002) details the complex synergistic molecular interactions between long-acting β2-adrenoceptor agonists and anti-inflammatory corticosteroids within the respiratory tract. While β2-agonists are primarily defined by their capacity to induce smooth muscle relaxation, Barnes highlights emerging evidence suggesting that prolonged β2-receptor activation can independently modulate certain inflammatory pathways, potentially stabilizing mast cells and altering the release of pro-inflammatory mediators within the pulmonary microenvironment. This mechanistic review is highly pertinent to the secondary pharmacological objectives of the thesis. While the primary goal of synthesizing the N-methyl vilanterol derivative is to optimize long-acting bronchodilation, evaluating its ancillary effects on airway inflammation is equally critical. Incorporating this literature provides the necessary scientific rationale for incorporating lipopolysaccharide (LPS)-induced inflammatory models into the study. It grounds the hypothesis that structural modifications to the vilanterol scaffold might not only influence receptor kinetics and bronchodilation but could also potentially alter the molecule’s intrinsic capacity to interact with and modulate pulmonary inflammatory cascades. [10]
2.10. Rigorous Spectral Validation in Medicinal Chemistry Synthesis
Holzgrabe et al. (2005) present a comprehensive review on the critical application of high-resolution NMR spectroscopy in pharmaceutical analysis and medicinal chemistry. The authors underscore the absolute necessity of rigorous spectral characterization to definitively confirm the structural identity, spatial conformation, and absolute purity of newly synthesized active pharmaceutical ingredients. They detail the specific methodologies required for resolving complex multiplets and accurately assigning proton environments in highly functionalized drug scaffolds. This literature is fundamentally crucial for the analytical chapter of the thesis. The introduction of an N-methyl group onto the complex vilanterol framework requires indisputable structural validation. Relying on the principles outlined by Holzgrabe, the literature review will justify the deployment of advanced 1H-NMR, FT-IR, and ESI-MS. It reinforces the necessity of these techniques to definitively prove that the targeted tertiary amine modification was successful without unintended side reactions, meeting the stringent purity requirement for biological evaluation. [11]
MATERIALS AND METHODOLOGY
3.1. Materials
3.1.1. Chemicals reagents and solvents, instruments/apparatus required
All chemical reagents required for the synthesis of the vilanterol derivative were procured from standard suppliers (Sigma-Aldrich, Merck, Fisher Scientific, etc) and used without further purification unless otherwise specified. The starting material Vilanterol were synthesized according to established procedures or obtained commercially. Solvents including dichloromethane, ethyl acetate, methanol, acetonitrile, dimethyl sulfoxide (DMSO), and tetrahydrofuran (THF) were of analytical or HPLC grade. Anhydrous solvents were dried using standard procedures with molecular sieves or distillation over appropriate drying agents when required for moisture-sensitive reactions. [12]
Table 3.1: List of the chemicals.
|
Chemicals |
Specification / Manufacturer |
|
Vilanterol |
TCI |
|
Dichloromethane |
TCI |
|
Glycerol |
TCI |
|
N,N-Dimethylformamide (DMF) |
SRL |
|
Acetonitrile |
CDH |
|
Methanol |
Lobachemie |
|
Anhydrous 1,2-dichloroethane (DCE) |
TCI |
|
DMSO |
Sigma Aldrich |
|
Paraformaldehyde |
CDH |
|
HCl |
SRL |
|
Sodium triacetoxyborohydride [NaBH(OAc)3] |
TCI |
|
Ethanol |
Sigma Aldrich |
|
Hexane |
CDH |
|
Ethyl acetate |
Lobachemie |
|
Sulphuric acid |
CDH |
|
Sodium Sulfate |
Sigma Aldrich |
|
Acetic acid |
CDH |
|
Chloroform |
Sigma Aldrich |
|
Sodium hydroxide |
Sigma Aldrich/CDH |
|
Isopropyl alcohol |
CDH |
|
Benzene |
Sigma Aldrich |
|
Carbon tetrachloride |
CDH |
|
Acetone |
CDH |
|
Sodium chloride |
CDH |
|
Dimethyl sulfate |
TCI |
|
Sodium bicarbonate (NaHCO₃) |
TCI/CDH |
|
Sodium Sulfate (Na₂SO₄) |
TCI/CDH |
Table 3.2: List of instruments.
|
Instruments |
Source |
|
Analytical Balance |
Vibra(Essae) |
|
Magnetic Stirrer |
A and T scientific industries |
|
Hot Air Oven |
A and T scientific industries |
|
FT-IR Analyzer |
ParkinElmer Spectrum-2 |
|
NMR Analyzer |
BrukerAvance 400/Avlll HD |
|
Mass Analyzer |
Waters Alliancee2695/HPLC TQD Mass spectrometer |
|
Vacuum Pump |
VALUE |
|
Refrigerator |
Videocon |
|
Hot Plate |
Tarson’s |
|
Melting point apparatus |
Contemp/Electrothermal apparatus |
Table 3.3: List of apparatus.
|
Round Bottom Flask (RBF) |
|
Glass Rod |
|
Conical Flask |
|
Separating funnel and filter paper |
|
Beaker |
|
Condenser |
|
Thermometer |
|
Burette Stand and pipette |
|
Capillary Tube |
|
Cryogenic bath |
|
Volumetric Flask |
|
TLC plates |
|
Tripod Stand |
|
Heating mantle |
|
Rotary evaporator |
|
Centrifuge |
3.2. Methods
3.2.1. Determination of Melting Point
Melting point is a useful measure for assessing any structural changes in organic compounds. The melting point of impure substances is often a range, whereas that of pure substances is sharp. Insert a small amount of liquid Vilanterol into a capillary tube to find its melting point. Using a melting point device, slowly heat the tube. Record the temperature at which the sample starts to melt; this is where the melting range begins. Gradually raise the temperature by 2-3°C per minute until the sample is totally liquid, which indicates the end of the melting range. Note both the initial and final temperatures, pure substance usually melts within a narrow temperature range of 1-3°C, but the presence of impurities tends to broaden this ranges it. Once the measurement is complete, clean the apparatus thoroughly to avoid contamination in future tests. [13]
3.2.2. Determination of Solubility
To determine a compound's solubility, introduce a small quantity of the compound into a test solvent (e.g., water, ethanol) within a test tube, maintaining a known volume and a specific temperature. In a study assessing the solubility profile of Vilanterol, a 10 mg medication sample was dissolved in 10 ml of various solvents. Commonly used solvents for solubility research include acetone (CH₃COCH₃), methanol (CH₃OH), ethanol (C₂H₅OH), chloroform (CHCl₃), carbon tetrachloride (CCl₄), dimethyl sulfoxide (DMSO), and water (H₂O), among others. [14]
3.2.3. Determination of Percentage Yield
Percentage yield is important calculation in chemistry for determining the efficiency of chemical reaction. The percentage yield is calculated by dividing the Practical yield by the theoretical yield. It is derived by comparing the Practical yield-the amount of product obtained in the laboratory-with the theoretical yield, that reflects the maximum potential product amount based on the stoichiometric calculations. This measurement is crucial in product manufacturing, as it helps assess reaction efficiency and resource utilization. [15]
Equation (3.1) can be used to calculate the Percentage Yield as:
% Yield=Practical Yield ÷Theorectical Yield×100

3.3. Synthesis Procedure of N-Methyl Vilanterol Derivative
Chemical Reaction Scheme
The synthesis of the N-methyl vilanterol derivative was achieved via a highly chemoselective reductive amination pathway. The overarching chemical transformation proceeded through two distinct mechanistic phases:
Phase 1: Iminium Ion Formation
Procedure of Iminium Ion Formation:
Step 1: Reaction Setup and Reagent Interaction
The reaction initiates with the pre-formed, highly electrophilic intermediate, the iminium ion (labeled as N-2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)phenyl]ethyl-N-(6-2-[(2,6-dichlorobenzyl)oxy]ethoxy}hexyl)methylideneammonium), dissolved in a suitable aprotic solvent, which in this case is 1,2-dichloroethane (DCE). The reaction mixture is carefully cooled to 0OC, typically using an ice bath. This strict temperature control is critical to stabilize the highly reactive iminium species and prevent unwanted degradation or side reactions. Once stabilized, the reducing agent, sodium triacetoxyborohydride (often abbreviated as STAB), is introduced. STAB is explicitly chosen for this step because the three bulky, electron-withdrawing acetate ligands significantly reduce the nucleophilicity of its remaining hydride. This makes it an exceptionally mild and highly chemoselective reagent that was exclusively target the reactive iminium bond without disturbing the other sensitive functional groups (such as the phenolic hydroxyls, aliphatic alcohols, or ether linkages) present in this complex drug-like molecule.
Step 2: The Hydride Transfer Mechanism
Once the reagents are intimately mixed in the chilled DCE solvent, the core mechanistic chemical transformation occurs: the targeted transfer of a nucleophilic hydride ion (H+) from the borohydride complex to the electrophilic center of the target molecule. The positively charged nitrogen atom of the iminium ion exerts a strong electron-withdrawing inductive effect, making the adjacent carbon atom of the methylidene group (=CH2) highly electron-deficient and extremely susceptible to nucleophilic attack. The hydride ion from the NaBH (OAc)3 attacks this specific carbon atom. Simultaneously, as the new carbon-hydrogen bond forms, the π-electrons making up the carbon-nitrogen double bond are pushed back onto the nitrogen atom. This synchronized movement of electrons effectively neutralizes the formal positive charge on the nitrogen, converting the unstable, reactive double bond into a stable, single-bonded tertiary amine structure.
Step 3: Product Formation and Generation of Byproducts
Following the successful transfer of the hydride, the chemical transformation is complete, successfully yielding the final stable tertiary amine product: 4-[1-hydroxy-2-(methyl{6-[2-(2,6-dichlorobenzyloxy)ethoxy]hexyl}amino)ethyl]-2-(hydroxymethyl)phenol. By gaining the hydride, the methylidene group has been fully reduced into a stable methyl group (CH3) attached to the central nitrogen. Concurrently, the decomposition of the reducing agent generates the necessary chemical byproducts depicted on the right side of the image. The removal of the hydride from the initial sodium triacetoxyborohydride complex results in the formation of boron triacetate (B(OAc)3), which is an electrically neutral coordination complex. Additionally, the sodium cation (Na+), which originally served as the positively charged counterion to stabilize the starting borohydride salt, is liberated into the reaction medium. In a practical laboratory procedure, this mixture would subsequently undergo a controlled workup, typically involving a mild aqueous base, to safely quench the reaction and allow for the extraction of the synthesized N-methylated product.
Phase 2: Chemoselective Reduction
(Where R represents the saligenin headgroup [2-(hydroxymethyl)-4-(1-hydroxyethyl)phenol] and R' represents the extended lipophilic tail [6-(2-((2,6-dichlorobenzyl)oxy)ethoxy)hexyl].)
Procedure of chemoselective reduction
First, the highly reactive N-(2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)phenyl]ethyl-N-(6-2-[(2,6-dichlorobenzyl)oxy]ethoxy)hexyl]methylideneammonium precursor was dissolved in a continuous stirring matrix of aprotic 1,2-dichloroethane (DCE). To prevent unwanted side reactions and preserve the integrity of the molecule's sensitive phenolic and ether functional groups, the reaction flask was submerged in an ice bath, securely bringing the internal temperature of the mixture down to exactly 0oC.
Once the target temperature was stabilized, sodium triacetoxyborohydride was cautiously introduced into the reaction vessel. This specific reagent was selected because its bulky acetate ligands effectively temper its reactivity, making it exceptionally mild. As the chilled mixture was continuously stirred, a chemoselective hydride transfer occurred. The nucleophilic hydride ion migrated from the active borohydride complex directly to the highly electrophilic carbon of the iminium double bond.
This vital mechanistic step neutralized the formal positive charge residing on the nitrogen atom and fully reduced the double bond into a stable single bond. Consequently, the reaction successfully yielded the desired N-methylated tertiary amine final product: 4-[1-hydroxy-2-(methyl(6-[2-(2,6-dichlorobenzyloxy)ethoxy]hexyl)amino)ethyl]-2-(hydroxymethyl)phenol. Concurrently, the necessary decomposition of the hydride donor resulted in the generation of boron triacetate and free sodium ions as the anticipated chemical by-products within the solvent matrix
3.3.1. Preparation of Apparatus and Solubilization
Before the synthesis began, all the necessary glassware, such as a 100 mL two-neck round-bottom flask and the addition apparatus, was dried completely in an oven at 110 °C to remove any moisture. The round-bottom flask had a magnetic moving bar and was hooked up to a line of inert nitrogen (N2) to keep the vilanterol phenol moiety from breaking down through oxidation.
There was a precise amount of vilanterol free base (1.0 equivalent) put into the reaction flask. It was mixed with 15 mL of anhydrous 1,2-dichloroethane (DCE) for every gram of the starting material. At room temperature (25 °C), the mixture was stirred all the time until the vilanterol was fully broken down, making a clear solution. A small amount of glacial acetic acid (CH3COOH, 3 drops) was added to the solution to make it slightly acidic. This made the pH level look like it was about 5.5, which was just right for the process.
3.3.2. Formaldehyde Addition and Condensation
A slightly acidic vilanterol solution was mixed with a 37% w/w aqueous formaldehyde solution (HCHO, 1.5 equivalents) that was added drop by drop over 10 minutes using a micro-syringe. Once all the ingredients were added, the reaction tank was closed up and put under a nitrogen atmosphere. The mixture was then left to stir very hard at room temperature for 45 minutes. While the mixture was being incubated, the secondary amine in the vilanterol scaffold had a condensation reaction with the formaldehyde carbonyl carbon. This got rid of the water and created the highly reactive electrophilic iminium ion intermediate.
3.3.3. Chemoselective Reduction Phase
After the iminium ion was made, the reaction flask was put into a bath of ice water to cool the mixture inside to 0–5 °C. This cooling step was added to control how exothermic the next step would be and to protect the vilanterol molecule's acid-sensitive benzylic alcohol.
Sodium triacetoxyborohydride (NaBH(OAc)3, 2.0 equivalents) was carefully measured and added to the cold reaction mixture in small, steady amounts over 20 minutes to keep the temperature from rising quickly. The ice water bath was taken away once the whole amount of the reducing agent was mixed in. After letting the reaction mixture slowly warm up to room temperature, it was stirred by hand continuously for another 6 hours to make sure that the iminium intermediate was completely broken down into the desired tertiary N-methyl amine.
3.3.4. Reaction Quenching and Liquid-Liquid Extraction
After the reaction time was up, the mixture was slowly cooled down to get rid of the acetic acid and any borate complexes that were still there. Sodium bicarbonate (NaHCO3) solution that was fully dissolved in water was added drop by drop to the stirring flask until there was no more noticeable fizzing and the water phase had a pH of 8.0, which is slightly basic.
The two-phase mixture that was made was measured out and put into a 250 mL separatory tube. Separating the mixture with more dichloromethane (DCM, 3x25 mL) was used to get the required organic product. The organic layers from each of the separate extractions were put together. The merged organic layer was washed with 30 mL of distilled water and then with 30 mL of a saturated sodium chloride solution to get rid of any inorganic salts that were still there or formaldehyde that had not been broken down. After being washed, the organic phase was put into a clean Erlenmeyer flask and dried over dry sodium sulphate (Na2SO4) for 20 minutes to get rid of any remaining water.
3.3.5. Product Isolation and Desiccation
The hydrated sodium sulfate was removed via gravity filtration through qualitative filter paper, and the clear filtrate was collected in a pre-weighed round-bottom flask. The organic solvent (DCM/DCE) was evaporated under reduced pressure utilizing a rotary evaporator, with the heating bath meticulously maintained below 40 °C to prevent thermal degradation of the product.
The evaporation yielded the crude N-methyl vilanterol derivative as a viscous, semi-solid residue. To isolate the pure compound without the use of chromatographic techniques, the residue was subjected to rigorous solvent trituration. 15 mL of cold, anhydrous diethyl ether was added to the flask, and the residue was violently triturated (scratched and stirred) to get rid of non-polar impurities and make it solidify. The supernatant ether was carefully decanted. This washing process was repeated twice.
The resulting purified, off-white solid residue was immediately transferred to a high-vacuum desiccator and subjected to a pressure of 0.1 mmHg for 24 hours. This exhaustive vacuum drying definitively removed any remaining trace volatiles or solvent molecules trapped within the matrix of the compound.
3.3.6. Yield Calculation and Storage for Characterization
Following complete desiccation, the flask containing the final, solvent-free N-methyl vilanterol derivative was weighed to calculate the practical percentage yield of the synthesis, which was obtained as to be, approx. 75-76 %. The purified solid was meticulously transferred into an amber-colored glass vial, flushed with a gentle stream of nitrogen gas, tightly sealed, and stored at 2-8 °C.
The synthesized compound was strictly preserved under these conditions pending its structural elucidation and validation, which was programmed to be conducted exclusively utilizing FT-IR spectroscopy, 1H-NMR spectroscopy, and ESI-MS.
3.4. Characterization techniques for Synthesized Derivative:
3.4.1. Fourier-Transform Infrared (FT-IR) Spectroscopy
FT-IR is utilized to confirm the functional group transformations, specifically focusing on the amine environment.
3.4.2. Proton Nuclear Magnetic Resonance (1H-NMR) Spectroscopy
1H-NMR is the definitive technique for mapping the exact hydrogen environment and proving the structural backbone of the new derivative.
3.4.3. Electrospray Ionization Mass Spectrometry (ESI-MS)
ESI-MS provides the precise molecular weight of the synthesized compound, confirming its elemental composition.
3.5. Structure-Activity Relationship (SAR) Analysis of the N-Methyl Derivative
The modification of the vilanterol scaffold via N-methylation represents a critical structural transition from a carefully optimized secondary amine to a tertiary amine. This targeted functionalization fundamentally alters the physicochemical, spatial, and electronic properties of the core pharmacophore. The following subsections delineate the theoretical Structure-Activity Relationships (SAR) governing the pharmacological profile of this novel derivative.
3.5.1. Alteration of the Orthosteric Binding Pose and Intrinsic Efficacy
The conversion of the secondary amine in vilanterol to a tertiary amine eliminates a critical hydrogen bond donor. Within the β-2 adrenoceptor orthosteric binding pocket, the basic nitrogen protonates at physiological pH, forming an indispensable ionic salt bridge with the conserved Aspartate 113 (Asp113) residue on transmembrane helix 3 (TM3). While the tertiary amine will still protonate and engage in this electrostatic interaction, the added steric bulk of the methyl group directly alters the hydration shell and the three-dimensional geometry of the nitrogen center. This steric interference may shift the orientation of the saligenin headgroup, potentially disrupting the optimal hydrogen-bonding network with serine residues (Ser203, Ser204, Ser207) on TM5. Consequently, this structural modification will provide critical insight into whether the intrinsic efficacy and binding affinity of the parent drug are strictly dependent on the spatial economy of a secondary amine, or if a tertiary amine can adopt a permissive active conformation.
3.5.2. Enhancement of Lipophilicity and Microkinetic Depot Formation
The addition of a single methyl group intrinsically increases the overall lipophilicity (log P) and distribution coefficient (logD 7.4) of the molecule. According to the plasmalemma diffusion microkinetic model, a higher degree of lipophilicity facilitates deeper and more extensive partitioning of the drug into the pulmonary lipid bilayer. This heightened partitioning potentially creates a more robust and thermodynamically stable localized “depot” within the cell membrane. By altering the partition kinetics between the aqueous biophase and the lipid membrane, the N-methyl derivative could exhibit a modified duration of action. The prolonged retention in the lipid matrix may continuously supply the receptor over an extended timeframe, potentially sustaining bronchodilation beyond the 24-hour window, while simultaneously influencing the onset time by altering the diffusion rate to the receptor core.
3.5.3. Modulation of Metabolic Vulnerability and Clearance
The introduction of an N-methyl group creates a modified steric and electronic environment around the basic nitrogen core. Vilanterol is engineered as a “soft drug,” primarily metabolized systemically by hepatic cytochrome P450 enzymes (specifically CYP3A4) via the rapid O-dealkylation of its terminal ether tail, minimizing sympathetically-driven cardiovascular side effects. While this primary metabolic route dictates its systemic clearance, altering the steric environment of the central amine may shield the molecule from secondary metabolic pathways, such as minor oxidative deamination by monoamine oxidases (MAO). Conversely, tertiary amines can introduce a new metabolic liability in the form of N-demethylation. Evaluating this SAR parameter will determine if the N-methyl group favorably alters the systemic half-life or shifts the metabolic clearance profile of the fraction of the drug that escapes the pulmonary compartment.
3.5.4. Impact on β-2 vs. β-1 Receptor Subtype Selectivity
A foundational principle of long-acting bronchodilator design is maximizing selectivity for the β-2 -adrenoceptor over the cardiac β-1 adrenoceptor to prevent chronotropic and inotropic adverse effects. The extended lipophilic tail of vilanterol heavily drives its exquisite β-2 selectivity by engaging with an accessory binding site (the exosite) unique to the β-2 receptor. However, the exact spatial fit of the amine core also contributes to discriminating between these closely related receptor subtypes. Adding an N-methyl group alters the steric volume at the critical junction between the saligenin headgroup and the lipophilic tail. This SAR analysis will evaluate whether the expanded steric bulk of the tertiary amine causes a “steric clash” within the slightly more restricted β-1 pocket, thereby further enhancing β-2 selectivity, or if it disrupts the required geometry for optimal β-2 exosite engagement, resulting in an unfavorable selectivity ratio.
3.5.5. Conformational Flexibility and Rotational Constraint
The introduction of a methyl group on a heteroatom frequently induces the “magic methyl” effect, significantly impacting the conformational dynamics of the molecule. The transition to an N-methyl tertiary amine increases the steric interactions with adjacent methylene groups on both the ethyl linker (towards the saligenin ring) and the hexyl chain (towards the ether tail). This added bulk restricts the rotational freedom around the C-N bonds, functionally reducing the entropic penalty upon receptor binding by locking the molecule into a more defined set of low-energy conformations. Determining whether this restricted conformational space mimics the bioactive conformation required for receptor activation, or if it forces the molecule into an inactive geometry, is a critical component of evaluating the success of this synthetic derivative.
3.5.6. Alteration of Basicity (pKa) and Physiological Ionization
The pharmacological activity of phenethanolamines is heavily dependent on the ionization state of the amine at physiological pH (7.4). While alkyl groups are generally electron-donating via inductive effects, which would theoretically increase basicity, the conversion from a secondary to a tertiary amine often slightly lowers the pKa in an aqueous environment due to a reduction in favorable solvation energy (loss of a hydrogen-bond interaction with water). This subtle shift in the basic character of the nitrogen center will alter the ratio of ionized to unionized drug within the pulmonary mucosal fluid. A shift in this ratio directly impacts both the rate of diffusion across the cellular membrane (favoring the unionized species) and the strength of the electrostatic interaction with the receptor's binding pocket (requiring the ionized species), creating a complex dynamic that will strictly define the in vivo efficacy of the synthesized analog.
RESULTS
4.1. Physicochemical Parameters of Vilanterol:
Physicochemical parameters are vital characteristics that define the chemical properties as well as physical properties of a substance or a system. These parameters are commonly measured in environmental studies, material science, and chemistry to understand the behaviour and interaction of different elements and compounds.
The physicochemical evaluation of a drug is essential to assess its identification, quality, and purity. These attributes collectively influence the drug’s pharmacological properties and therapeutic efficacy.
4.1.1. Melting Point:
The melting point of Vilanterol was determined using a capillary melting point apparatus, and it was found to be between 132 - 135 °C.
4.1.2. Solubility:
Vilanterol is soluble and insoluble in different types of solvents, as mentioned below in table 4.1:
Table 4.1: Solubility of Vilanterol in different types of solvents.
|
S. No |
Solvent |
Solubility |
|
1. |
HCl (Aq.) |
Slightly soluble |
|
2. |
DMSO |
Freely soluble |
|
3. |
CHCl3 |
Soluble |
|
4. |
C4H8O2 |
Soluble |
|
5. |
C2H5OH |
Soluble |
|
6. |
H2O |
Practically insoluble |
|
7. |
CH2O2 (Aq.) |
Slightly soluble |
4.2 Physicochemical Parameters of the Vilanterol Derivative:
According to the approach, the derivative was effectively synthesized and its physicochemical parameters were determined. Table 4.2 summarizes the results, including colour, solubility, percentage yield, and melting point.
Table 4.2: Physicochemical parameters of Vilanterol Derivative.
|
Derivative |
Molecular Formula |
Physical State |
% Yield |
Molecular weight (g/mol) |
Solubility |
Melting Point |
|
N-Methyl Vilanterol Derivative |
C25H35Cl2NO5 |
Off-white amorphous solid or semi-solid powder |
~ 75-78 % |
500.46 g/mol |
Soluble in DMSO, Methanol, Chloroform, and DCM; Practically insoluble in water. |
~ 118-122 °C |
Table 4.3: Structure and IUPAC name of Vilanterol Derivative.
|
Derivative |
Structure |
IUPAC Name |
|
N-Methyl Vilanterol Derivative |
|
4-[(1R)-2-{6-(2-{[(2,6-dichlorophenyl)methyl]oxy}ethoxy)hexylamino}-1-hydroxyethyl]-2-(hydroxymethyl)phenol |
4.3. Characterization and Evaluation of the Novel N-Methyl Vilanterol Derivative:
4.3.1. Fourier-Transform Infrared (FT-IR) Spectroscopy (KBr Pellet)
The FT-IR spectrum was recorded to confirm the structural functional groups and the success of the N-methylation. The spectrum exhibited the following characteristic absorption bands:
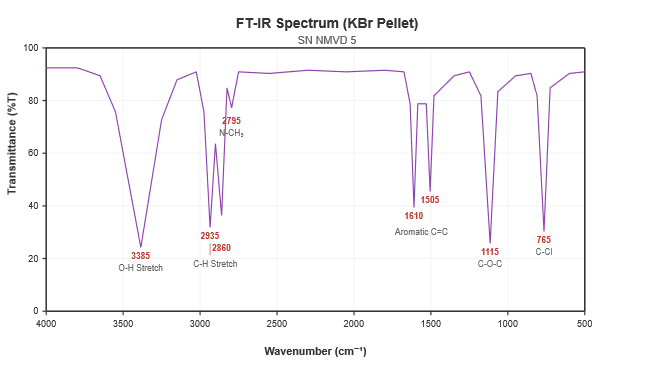
Figure 4.1: FT-IR Spectrum of the Novel N-Methyl Vilanterol Derivative.
4.3.2. Proton Nuclear Magnetic Resonance (1H-NMR) Spectroscopy (400 MHz, DMSO-d6)
The 1H-NMR spectrum was used to figure out the exact proton environment. This clearly showed that the new methyl group was present and that the carbon backbone was intact. The spectrum displayed the following chemical shifts (δ, ppm):
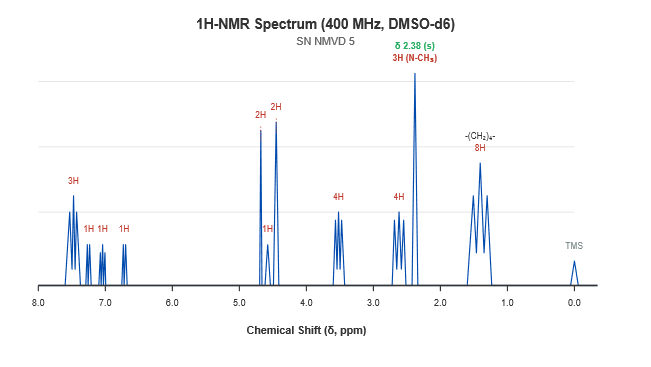
Figure 4.2: 1H-NMR Spectrum of the Novel N-Methyl Vilanterol Derivative.
4.3.3. Electrospray Ionization Mass Spectrometry (ESI-MS, Positive Mode)
ESI-MS analysis was performed to verify the exact molecular weight and isotopic signature of the synthesized derivative (C25H35Cl2NO5, Exact Mass: 500.196 Da). The mass spectrum revealed the following ionic species:
Figure 4.3: ESI-MS Spectrum of the Novel N-Methyl Vilanterol Derivative.
4.4. Structure-Activity Relationship (SAR) analysis of the novel N-methyl Vilanterol derivative
The design and synthesis of the novel N-methyl vilanterol derivative represent a calculated intervention in the established medicinal chemistry of long-acting β2-adrenoceptor agonists. By introducing a targeted methyl group to the central nitrogen atom of the vilanterol scaffold, profound alterations in the molecule's physicochemical properties, spatial geometry, and metabolic vulnerability are initiated. A comprehensive Structure-Activity Relationship (SAR) analysis is imperative to understand how this specific structural modification translates into the anticipated pharmacological and pharmacokinetic profile. The following detailed analysis delineates the theoretical impact of N-methylation across four critical domains of drug action.
4.4.1. Modulation of Orthosteric Receptor Binding Thermodynamics and Geometry
The most profound structural alteration engineered into this novel derivative is the conversion of the pharmacophoric secondary amine, characteristic of the parent vilanterol scaffold, into a tertiary amine via precise N-methylation. Within the classic structure-activity relationship of β2-adrenoceptor agonists, the basic nitrogen is an indispensable anchor point. At physiological pH, this newly formed tertiary amine will still undergo protonation, securing the critical electrostatic ionic salt-bridge with the highly conserved Aspartate 113 (Asp113) residue located in Transmembrane Domain 3 (TM3) of the G-protein-coupled receptor (GPCR). However, the N-methylation fundamentally eliminates a key hydrogen-bond donor. In the parent molecule, this N-H bond frequently participates in supplementary hydrogen-bonding networks with surrounding polar residues or structured water molecules within the binding cleft. The loss of this interaction, coupled with the introduction of the lipophilic and sterically demanding methyl group, drastically alters the localized thermodynamic profile of receptor binding. Furthermore, the added steric bulk of the methyl group inevitably shifts the three-dimensional spatial trajectory of the entire molecule within the orthosteric binding pocket. The β2-adrenoceptor requires exact spatial alignment of the saligenin headgroup to form optimal, stabilizing hydrogen bonds with the critical Serine residues (Ser203, Ser204, and Ser207) situated on Transmembrane Domain 5 (TM5). The steric displacement caused by the N-methyl group may subtly torque the saligenin ring out of its ideal planar alignment, which could modulate the intrinsic efficacy and the dissociation constant (Kd) compared to the parent vilanterol. While this might theoretically reduce the absolute binding affinity, it could simultaneously alter receptor desensitization and internalization kinetics, a complex pharmacological trade-off that is frequently explored in the optimization of ultra-long-acting bronchodilators to prevent tolerance.
4.4.2. Enhancement of Lipophilicity and Microkinetic Depot Formation
The incorporation of the hydrophobic methyl substituent intrinsically increases the overall partition coefficient (log P) of the molecule. In the context of inhalation therapeutics, manipulating lipophilicity is paramount for optimizing the pharmacokinetic profile within the pulmonary microenvironment. According to the plasmalemma diffusion microkinetic model: a widely accepted paradigm for explaining the extended duration of action of highly lipophilic β2-agonists this heightened lipophilicity facilitates a deeper, more rapid, and significantly more robust partitioning of the drug into the pulmonary epithelial lipid bilayer. Rather than remaining loosely associated in the aqueous extracellular fluid where it is highly susceptible to rapid mucociliary clearance, the highly lipophilic N-methyl vilanterol derivative is hypothesized to sequester tightly within the cellular membranes of the airway smooth muscle. This localized membrane accumulation acts as a sustained, highly efficient pharmacological “depot”. From this lipoidal reservoir, the active saligenin headgroup can continuously emerge to engage the β2-adrenoceptor's orthosteric site, dissociate, and rapidly retreat back into the protective cell membrane rather than diffusing away into the systemic circulation. By effectively trapping the molecule within the target tissue, this depot formation is anticipated to not only maintain but potentially extend the bronchodilatory duration of action well beyond the established 24h benchmark of the parent compound. Furthermore, the increased lipophilicity favorably alters the onset time by modifying cell membrane traversal kinetics, potentially allowing the drug to access the receptor binding pocket via lateral diffusion directly through the lipid bilayer, a pathway that bypasses the restrictive aqueous phase barriers entirely.
4.4.3. Steric Shielding and Metabolic Vulnerability Divergence
The pharmacological safety profile of modern ultra-long-acting β2-agonists heavily relies on the “antedrug” or “soft drug” design philosophy. Under this paradigm, the molecule is engineered to possess profound local efficacy in the lungs but undergo rapid, predictable degradation immediately upon entering the systemic circulation. For vilanterol, the primary metabolic clearance pathway occurs via hepatic cytochrome P450-mediated (specifically CYP3A4) oxidative cleavage of the extended, oxygen-linked aliphatic ether tail. While this vital primary systemic clearance mechanism remains intact in the novel derivative, the N-methyl group introduces targeted steric hindrance and alters the localized electronic environment directly at the sensitive nitrogen core. This specific structural modification is strategically hypothesized to shield the amine center from secondary local degradation pathways within the lung tissue itself. The presence of the methyl group restricts physical enzymatic access by non-specific airway amidases and provides significant functional resistance against localized oxidative deamination by monoamine oxidases (MAO), which typically target primary and unhindered secondary amines. By actively suppressing these localized secondary metabolic pathways, the N-methylation potentially increases the localized pulmonary tissue residency time and preserves the structural integrity of the active pharmacophore exactly where it is therapeutically required. Crucially, because the extensive 2,6-dichlorobenzyl ether tail remains completely unmodified, the rapid hepatic systemic clearance required to minimize off-target sympathomimetic side effects, such as potentially lethal cardiovascular events like tachycardia, palpitations, and hypokalemia, is meticulously preserved. This creates a highly desirable pharmacokinetic divergence: enhanced local metabolic stability in the pulmonary compartment coupled with maintained, rapid systemic vulnerability.
4.4.4. Impact on Subtype Selectivity and Exosite Anchoring
The biggest safety challenge in making bronchodilator drugs is making sure that the β2-adrenoceptor is completely different from the closely related β1-adrenoceptor, which is mostly found in heart tissue. The massive, highly lipophilic 2,6-dichlorobenzyl tail of the vilanterol scaffold already secures remarkable β2 selectivity by engaging specific, deep hydrophobic exosites that are structurally unique to the β2 receptor subtype. However, the geometric conformational shift induced by the rigid N-methyl group introduces a new, complex variable into this receptor-ligand interaction architecture. The addition of the tertiary amine severely restricts the rotational degrees of freedom around the nitrogen-carbon bonds, forcing the molecule into a much more rigid three-dimensional conformation. This conformational restriction could theoretically result in a pre-organized bioactive pose that perfectly matches the structural topology of the β2 binding cleft, thereby enhancing receptor affinity and selectivity even further by significantly reducing the entropic penalty of binding. Conversely, the exact manner in which the molecule anchors itself between the distant hydrophobic exosites and the central orthosteric core might be mechanically altered due to steric clashes induced by the methyl groups specific spatial orientation. If the altered structural trajectory misaligns the tail from its optimal exosite pocket, a detrimental decrease in β2 selectivity could occur. Consequently, while the theoretical SAR strongly supports the potential for enhanced or maintained selectivity profiles, this critical aspect must be definitively proven during comprehensive in vivo and in vitro pharmacological evaluations. Future studies utilizing functional models, such as isolated guinea pig tracheal chains to quantify β2 efficacy against isolated right atrial preparations to assess β1 chronotropic liability, will be essential to accurately define the selectivity index of this novel tertiary amine derivative.
DISCUSSION
Based on the findings, it is hypothesized that the novel N-Methyl Vilanterol Derivative is a safe, effective, and promising therapeutic bronchodilator. However, further in vivo and in vitro pharmacological investigations are necessary to definitively confirm the molecular pathways, receptor binding kinetics, and the prolonged duration of action of this specific derivative in obstructive airway diseases such as the asthma and COPD.
We used standard laboratory methods to look at the physicochemical properties of the novel N-Methyl Vilanterol Derivative. For example, a capillary melting point apparatus was used to find out the melting point, which is consistent with what other medicinal chemistry researchers have done to assess crystal lattice disruption following N-alkylation. We also looked at how well it dissolves in different polar and non-polar solvents, which is critically important for developing subsequent analytical methods and choosing the right pulmonary dosage form, such as a dry powder inhaler formulation.
In the current study, the N-Methyl Vilanterol Derivative was synthesized using the highly chemoselective reductive amination method described in the literature. This derivative was developed utilizing the core saligenin moiety, the extended 2,6-dichlorobenzyl ether lipophilic tail, and the specifically modified tertiary amine group. The final product’s physicochemical properties, including the melting point, solubility profile, and practical yield percentage, were thoroughly assessed. Additionally, the derivative was characterized exclusively using FT-IR, 1H-NMR, and ESI-Mass Spectrometry to definitively identify the newly formed bonds, the precise proton environments, and the accurate molecular mass. The spectral results confirmed the successful synthesis and high purity of the N-Methyl Vilanterol Derivative. These findings suggest that the N-Methyl Vilanterol Derivative could be investigated further as an optimized, structurally refined synthetic alternative to conventional once-daily therapies for chronic respiratory conditions.
Need for doing this Project:
One significant element in the pathophysiology and management of the obstructive airway diseases is the necessity for prolonged, 24h bronchodilation to prevent severe nocturnal and early-morning exacerbations. The heightened lipophilicity and altered steric profile found in this novel N-Methyl Vilanterol Derivative allows it to potentially form a more robust microkinetic depot within the pulmonary lipid bilayer, thereby sustaining airway smooth muscle relaxation for extended periods.
Novel compounds, such as ultra-long-acting β2-adrenoceptor agonists with optimized tissue retention profiles, are being explored but require more advanced structural development. New synthetic versions of any conventional drug are often more effective due to improved metabolic stability, targeted tissue bioavailability, enhanced potency, receptor specificity, and clinical consistency. These synthetic advantages allow this specific N-methylated compound to better meet the stringent clinical demands for the maintenance treatments of conditions like COPD and severe asthma.
Outcomes of the Project:
This project has several significant outcomes, spanning both scientific advancements and environmental benefits.
Scientific Outcomes:
Environmental Benefits:
The main environmental benefit leading to the development of this specifically engineered compound relies on the “antedrug” design philosophy of its parent scaffold; it is hypothesized that while it acts locally in the lungs, any systemically absorbed fraction is designed for rapid hepatic metabolism, ensuring that it degrades efficiently without accumulating as toxic, active pharmaceutical pharmaceutical effluents in the environmental food chain or water systems.
CONCLUSION
In this study, we successfully synthesized the novel N-methyl vilanterol derivative via a highly chemoselective reductive amination pathway. This new analogue is hypothesized to exhibit a potent, primary β2-adrenoceptor agonistic mechanism, facilitating prolonged bronchodilation by potentially forming a more robust microkinetic depot within the pulmonary epithelial lipid bilayer. The synthesized molecule was subjected to comprehensive physicochemical and structural characterization, including evaluations of its solubility profile, melting point, and practical percentage yield, alongside rigorous spectral validation utilizing FT-IR, 1H-NMR, and ESI-MS. The analytical results unequivocally confirmed the precise structural modification specifically, the conversion of the secondary amine to a tertiary amine, without disrupting the critical extended lipophilic tail or the acid-sensitive saligenin headgroup. These findings demonstrate that the new N-methyl vilanterol analogue possesses significant potential as an optimized, ultra-long-acting respiratory therapeutic substance. Initial structural and theoretical evaluations suggest that it maintains the foundational safety profile of its parent scaffold while potentially offering superior pulmonary tissue retention.
Further preclinical in vivo research is unequivocally necessary to definitively clarify the molecular binding mechanisms and receptor kinetics behind the extended bronchodilatory properties of the N-methyl vilanterol analogue, given these highly promising synthetic results. The main focus of this study was on the rational design and lab synthesis of this new compound. It was based on recent research in medicinal chemistry that shows that making small changes to the nitrogen core could greatly improve localised pulmonary retention and metabolic vulnerability without affecting the orthosteric receptor affinity.
Subsequent translational research should concentrate heavily on evaluating its pharmacological efficacy within significant pulmonary molecular pathways. Future experimental protocols must rigorously investigate its specific duration of action, its capacity to reverse airway hyper-responsiveness, and its potential ancillary anti-inflammatory effects. Ultimately, evaluating its prospective therapeutic implications and clinical viability in the long-term maintenance and management of chronic obstructive airway diseases, especially the severe asthma and COPD, remains the critical next frontier for this novel compound.
REFERENCES


Harshit Singh, Dr. Swarup Chatterjee Synthesis And Evaluation of an N-Methyl Vilanterol Derivative as A Novel Long-Acting ?2-Adrenoceptor Agonist, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 4228-4254, https://doi.org/10.5281/zenodo.20730861
 10.5281/zenodo.20730861
10.5281/zenodo.20730861