
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, Sagar Institute of Technology and Management, Department of Pharmacy, Barabanki.
Breast cancer is one of the most common and deadly cancers that affect women globally. Because of its rising frequency, resistance to treatment, and side effects from traditional chemotherapy, it remains a significant global health concern. The search for novel anticancer agents possessing improved safety, selectivity, and additional pharmacological benefits has therefore become an important area of medicinal chemistry research. In the present investigation, a series of novel benzothiazole-based Schiff base derivatives were designed, synthesized, characterized, and evaluated for their anticancer and antioxidant potential. The target compounds (3a–3f) were synthesized through a multistep synthetic route involving preparation of substituted benzothiazol-2amine intermediates followed by condensation with cinnamaldehyde derivatives. The anticancer potential of the synthesized derivatives was assessed through molecular docking studies using AutoDock Vina against two important breast cancer–associated target proteins, namely Epidermal Growth Factor Receptor kinase domain (EGFR, PDB ID 2J6M) and Human Estrogen Receptor Alpha Ligand (ER?, PDB ID: 3ERT). Docking analysis demonstrated favorable binding affinity and stable interaction profiles of the synthesized compounds within the active binding cavity of both receptors. Several derivatives exhibited docking scores comparable to or higher than the standard drug Tamoxifen, indicating promising receptor binding potential.
Cancer is a complicated group of illnesses marked by the unchecked proliferation and dissemination of aberrant cells that can invade surrounding tissues and spread to other organs these biological hallmarks distinguish cancer from benign growths and make it a formidable public health threat worldwide.
Cancer is one of the most significant causes of death worldwide. According to the WHO, cancer was accountable for an estimated 10 million deaths in 2020, representing nearly one in six deaths globally.
Breast cancer is unique among cancer types because of its extremely high incidence and high fatality rate. Globally, an estimated 2.3 million women received a breast cancer diagnosis in 2022, and the disease claimed the lives of about 670,000 people. In 157 out of 185 nations, breast cancer was the most prevalent disease diagnosed in women. It can strike at any age after puberty, and the risk rises with age.[6]
Benzothiazole is an important bicyclic heterocyclic compound that has attracted considerable attention in medicinal and pharmaceutical chemistry due to its unique structural features and broad spectrum of biological activities. Structurally, benzothiazole consists of a fused benzene ring and a thiazole moiety containing both sulfur and nitrogen heteroatoms. This conjugated heteroaromatic framework confers remarkable chemical stability, planarity, and electronic versatility, making benzothiazole a privileged scaffold in drug discovery and development.
The presence of heteroatoms within the benzothiazole nucleus allows for diverse intermolecular interactions, including hydrogen bonding, π–π stacking, and coordination with biological targets.[39]
These properties enable benzothiazole derivatives to interact effectively with enzymes, receptors, nucleic acids, and other biomacromolecules, thereby contributing to their wide-ranging pharmacological potential.[40]
Benzothiazole-Based Agents in Role in Oncology and Cancer Diagnostics
Benzothiazole derivatives have gained considerable attention in oncology, both as therapeutic agents and diagnostic tools. Their planar aromatic nature facilitates interactions with DNA, enzymes, and signaling proteins involved in cancer cell proliferation and survival. Quizartinib (Vanflyta), a highly selective type II FLT3 inhibitor approved for the treatment of acute myeloid leukemia, exemplifies the potential of benzothiazole-related scaffolds in targeted cancer therapy. By selectively inhibiting aberrant kinase signaling, such compounds contribute to improved treatment specificity and reduced systemic toxicity. Phortress is an experimental benzothiazole-based antitumor prodrug designed to target breast, ovarian, and renal carcinomas. It undergoes metabolic activation within tumor cells, generating cytotoxic intermediates that selectively impair cancer cell viability. This strategy underscores the value of benzothiazole derivatives in prodrug design and tumor-selective therapy.[43]
Scheme
Materials
List of Chemical Used
Methodology
Molecular Docking Study
Molecular docking studies were performed to investigate the binding affinity, molecular interactions, and possible binding orientations of the synthesized benzothiazole derivatives (3a–f) against selected breast cancer–related target proteins. Docking analysis helps in understanding the interaction behavior of synthesized compounds within the active binding cavity of receptor proteins and predicts their potential biological activity at the molecular level.
Fig 4.1 Molecular Docking
The docking study performed using AutoDock Vina, which is widely used for predicting ligand–protein interactions based on binding free energy calculations and conformational flexibility. The synthesized derivatives were evaluated against two important breast cancer–associated molecular targets, namely the EGFR kinase domain and Human Estrogen Receptor Alpha.
Selection of Target Proteins
The current molecular docking study's target proteins were linked to the advancement of breast cancer and cellular proliferation. The Protein Data Bank (PDB) provided the proteins' three-dimensional crystal structures.
The selected target proteins were:
Target 1: EGFR kinase domain (PDB ID: 2J6M)
The Epidermal Growth Factor Receptor (EGFR) is a membrane-bound receptor possessing intrinsic tyrosine kinase activity and is critically involved in regulating several cellular processes such as proliferation, survival, differentiation, migration, and angiogenesis. Dysregulation or excessive expression of EGFR has been frequently observed in multiple malignancies, particularly breast cancer, where it contributes to uncontrolled tumor growth and metastatic progression. Due to its significant role in cancer cell signaling pathways, EGFR has emerged as an important molecular target for the development of anticancer agents aimed at suppressing tumor progression through inhibition of kinase activity.
Target 2: Human Estrogen Receptor Alpha Ligand (PDB ID: 3ERT)
Human Estrogen Receptor Alpha (ERα) is a ligand-activated nuclear transcription factor that plays a crucial role in the initiation and progression of hormone-responsive breast cancer. Upon interaction with estrogenic compounds, ERα regulates the expression of genes associated with cellular growth, proliferation, and survival. Abnormal activation of this receptor has been closely linked with estrogen-dependent breast tumor development. Consequently, targeting ERα through receptor antagonism or modulation has become a well-established therapeutic strategy in the management of estrogen receptor–positive breast cancer.
Ligand Preparation
The chemical structures of all synthesized benzothiazole derivatives (3a–f) were drawn using ChemDraw software. The two-dimensional structures were converted into three-dimensional conformations and subjected to geometry optimization in order to obtain energetically stable molecular structures suitable for docking analysis.
The optimized structures were saved in MOL format, then Open Babel was used to transform them into PDB format. Hydrogen atoms were added during ligand preparation, and energy minimization was used to eliminate undesirable conformations and steric hindrance. Ultimately, all ligand structures were transformed into PDBQT format, which is necessary for docking simulations using Auto Dock Vina.
Protein Preparation
The crystal structures of the selected proteins were downloaded in PDB format from the Protein Data Bank. Prior to docking, protein structures were prepared carefully to ensure proper ligand binding analysis.
Protein preparation involved the following steps:
Protein preparation done by AutoDock tools to obtain receptor files compatible with AutoDock Vina.
Active Site Identification
The active binding site of each target protein was identified based on the binding position of the co-crystallized ligand present in the crystal structure. The amino acid residues surrounding the ligand-binding cavity were selected for grid box generation.
For EGFR kinase domain (2J6M), important active site residues included Leu718, Val726, Ala743, Lys745, Thr790, Leu788, and Leu844, which are known to participate in ATP-binding and kinase inhibition.
For Human Estrogen Receptor Alpha (3ERT), important amino acid residues involved in ligand binding included Glu353, Arg394, His524, Leu387, and Phe404.
Docking Procedure
Docking simulations were performed using AutoDock Vina to evaluate the interaction of synthesized compounds with the selected receptor proteins. A grid box was generated around the active site region of each protein to allow ligand flexibility and proper exploration of the binding cavity.
During docking, the ligands were allowed to adopt different conformations and orientations inside the receptor binding pocket. AutoDock Vina calculated the binding affinity based on intermolecular interactions such as hydrogen bonding, hydrophobic interactions, van der Waals forces, and electrostatic interactions.
The docking score obtained from AutoDock Vina was expressed in kcal/mol. Lower binding energy values indicated stronger ligand–protein interaction and better binding stability. The best docking pose for each ligand was selected based on minimum binding energy and favorable interaction profile.
Post-Docking Analysis and Visualization
The docked ligand–protein complexes were analyzed for intermolecular interactions and binding orientation using PyMOL and BIOVIA Discovery Studio Visualizer.
Two-dimensional and three-dimensional interaction analyses were performed to identify:
The amino acid residues involved in ligand stabilization inside the active site were carefully examined and compared with the interactions shown by the standard drug.
Standard Drug for Comparative Docking
The docking results of synthesized compounds were compared with the standard anticancer drug Tamoxifen. Tamoxifen was selected as the reference ligand because of its established role in breast cancer therapy and its reported interaction with estrogen receptor and related oncogenic targets.
The comparative docking study helped in evaluating the potential anticancer activity of the synthesized benzothiazole derivatives and identifying compounds with promising receptor binding characteristics for further biological evaluation.
Synthetic Procedures
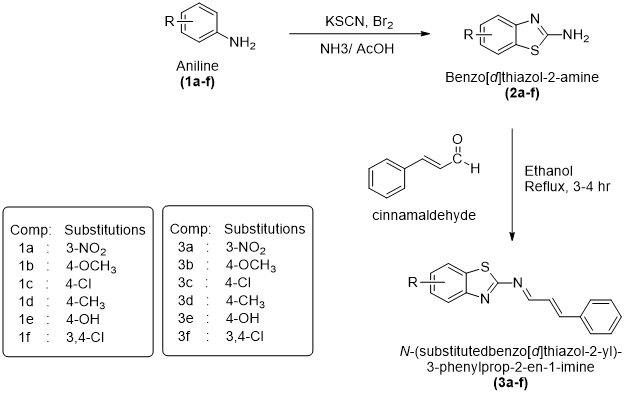
General Procedure for the Synthesis of Substituted Benzothiazol-2amine Derivatives (2a–f)
The substituted benzo[d]thiazol-2-amine derivatives (2a–f) were prepared by an oxidative cyclization method using suitably substituted aniline derivatives as starting materials in the presence of potassium thiocyanate under acidic conditions. In the initial step, potassium thiocyanate (8 g, 0.08 mol) along with the corresponding substituted aniline derivative (1.45 g, 0.01 mol) was dissolved in 20 mL of pre-chilled glacial acetic acid under constant stirring to obtain a homogeneous reaction mixture. The reaction temperature was carefully controlled at a low level with the help of an ice–salt bath in order to minimize excessive heat evolution and ensure smooth progression of the cyclization process.
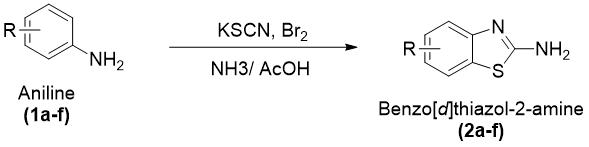
A bromine solution prepared by dissolving bromine (1.6 mL) in glacial acetic acid (6 mL) was added slowly to the reaction mixture in a dropwise manner over approximately 105 minutes with constant stirring. Careful addition of bromine was necessary to control the exothermic nature of the cyclization process. After complete addition, the reaction mixture was stirred continuously for an additional 2 hours under cold conditions and then allowed to stir at room temperature for nearly 10 hours. The reaction mixture was subsequently kept undisturbed overnight to ensure completion of cyclization.
After completion of the reaction, the mixture was heated at approximately 85 °C with the addition of 6 mL of distilled water and filtered while hot to obtain the first filtrate. The remaining residue was further extracted using 10 mL of glacial acetic acid at the same temperature and filtered again to obtain a second filtrate. Both filtrates were combined and allowed to cool to room temperature. The combined solution was then neutralized carefully using concentrated ammonia solution until the pH reached nearly 6, leading to the formation of a precipitated solid product.
The separated crude product was collected by filtration, washed appropriately, and subjected to purification using activated charcoal treatment. Final purification was carried out by recrystallization from a benzene–ethanol solvent mixture (1:1), affording pure substituted benzo[d]thiazol-2-amine derivatives (2a–f) in crystalline form.
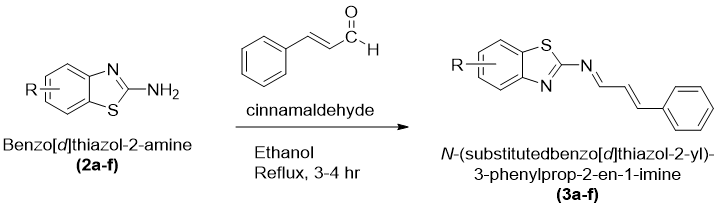
Synthesis of N-(Substitutedbenzo[d]thiazol-2-yl)-3-phenylprop-2-en-1-imine Derivatives (3a–f)
The Schiff base derivatives of benzothiazole, namely N-(substitutedbenzo[d]thiazol-2-yl)-3-phenylprop-2-en1-imine (3a–f), were synthesized by condensation of substituted benzo[d]thiazol-2-amines with cinnamaldehyde under reflux conditions. Equimolar quantities of substituted benzo[d]thiazol-2-amine and cinnamaldehyde were dissolved in absolute ethanol in a clean round-bottom flask equipped with a reflux condenser. To promote the condensation reaction and facilitate imine bond formation, a few drops of glacial acetic acid were introduced as a catalyst.
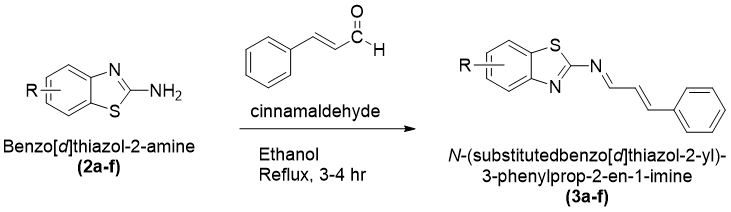
The reaction mixture was heated under reflux for approximately 3–4 hours with continuous stirring. The reaction progress is observed using TLC using a suitable mobile phase system. Formation of a new spot corresponding to the desired product confirmed completion of the reaction.
After completion, the reaction mixture was allowed to attain room temperature and was then transferred slowly into ice-cold distilled water with constant stirring. A colored solid precipitate separated out gradually, indicating formation of the Schiff base derivative. The obtained precipitate was collected by vacuum filtration and washed thoroughly with distilled water to remove traces of catalyst and any unreacted reactants. The crude products were dried under reduced pressure and subsequently purified by recrystallization using ethanol as the solvent. Pure crystalline derivatives of N-(substitutedbenzo[d]thiazol-2yl)-3phenylprop-2-en-1imine (3a–f) were obtained after recrystallization and preserved in airtight containers for further characterization and biological evaluation.
Purity Determination of Synthesized Compounds
Purity of synthesizd compounds was determine by perfoerming Thin Layer Chromatography and recrystallization technique.
Characterization of Synthesized Compounds
The synthesized benzothiazole derivatives were characterized using various physicochemical and spectral analytical techniques to confirm their identity, purity, and structural features. The characterization studies included melting point determination, FT-IR, 1H-NMRspectroscopy, mass spectral analysis, and elemental analysis. These analytical techniques provided detailed information regarding the structural framework, functional groups, molecular weight, and composition of the synthesized compounds.
Evaluation of Antioxidant Activity by DPPH Free Radical Scavenging Assay
The antioxidant potential of the selected synthesized benzothiazole derivatives was assessed by employing the DPPH free radical scavenging method. This assay is widely recognized for its simplicity, sensitivity, and reliability in determining the ability of compounds to donate hydrogen atoms or electrons for neutralization of free radicals. The method is based on reduction of the stable DPPH radical, which undergoes a visible color change upon interaction with antioxidant molecules.
DPPH is a stable nitrogen-centered free radical that exhibits a deep violet coloration in methanolic solution due to its strong absorption around 517 nm. When an antioxidant compound reacts with DPPH, the radical accepts a hydrogen atom or electron from the test compound and becomes reduced to a non-radical form. This reduction is accompanied by fading of the violet color to pale yellow, which can be quantitatively measured spectrophotometrically. The degree of discoloration reflects the radical scavenging efficiency of the tested compounds.
RESULTS & DISCUSSION
Molecular Docking Analysis of Synthesized Benzothiazole Derivatives Against EGFR Kinase Domain (PDB ID: 2J6M)
The synthesized derivatives (3a–3f) were subjected to molecular docking analysis against the EGFR kinase domain using the crystal structure of EGFR tyrosine kinase (PDB ID: 2J6M). The docking study was performed to evaluate the binding affinity and molecular interactions of the synthesized compounds within the active binding pocket of the receptor. The obtained docking scores were compared with the standard drug Tamoxifen.
The docking results revealed that all synthesized derivatives exhibited appreciable binding affinity toward the EGFR kinase domain, with binding energies ranging from −7.5 to −7.8 kcal/mol, which were comparable to or better than the standard drug tamoxifen (−7.4 kcal/mol). Among all tested compounds, derivative 3d demonstrated the highest binding affinity with a docking score of −7.8 kcal/mol, indicating strong interaction and favorable accommodation within the receptor active site.
Table: 2D Interaction and Binding Affinity of Benzothiazole Derivatives Against EGFR Kinase Domain (PDB ID: 2J6M)
|
Comp |
Binding Affinity (kcal/mol) |
H-Bond Interactions |
π–π / Hydrophobic Interactions |
2D Interaction |
|
3a |
-7.6 |
Leu788 |
Val726, Ala743, Leu844, Leu718 |
|
|
3b |
-7.5 |
- |
Leu718, Thr790, Ala743, Leu788, Leu844, Val726, Lys745 |
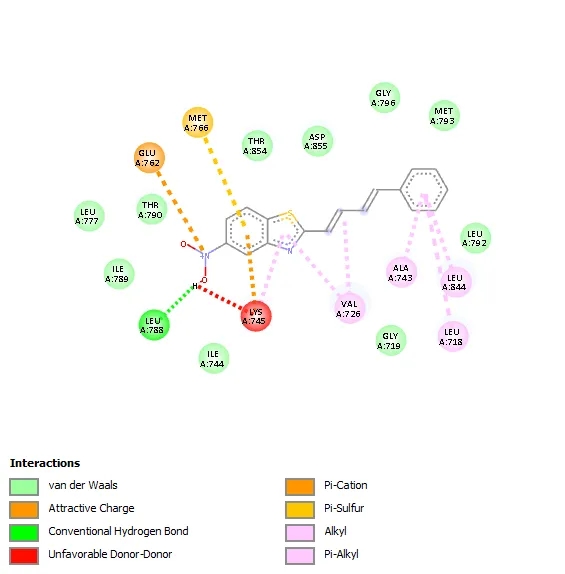
|
|
3c |
-7.7 |
- |
Leu718, Thr790, Ala743, Leu788, Leu844, Val726, Lys745 |
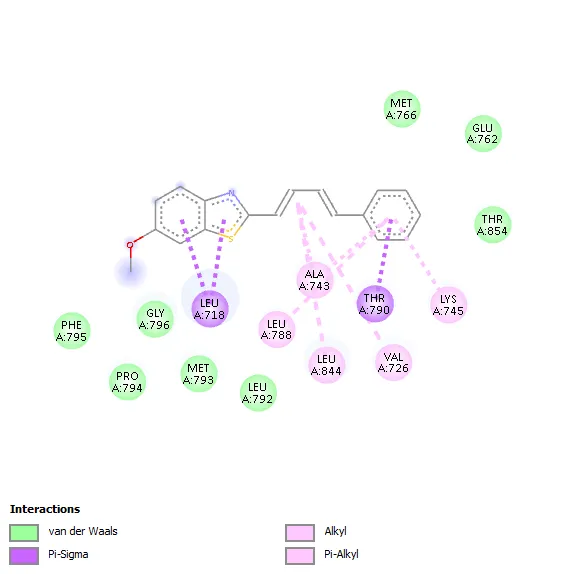
|
|
3d |
-7.8 |
- |
Leu718, Thr790, Leu788, Ala743, Leu844, Val726, Lys745 |
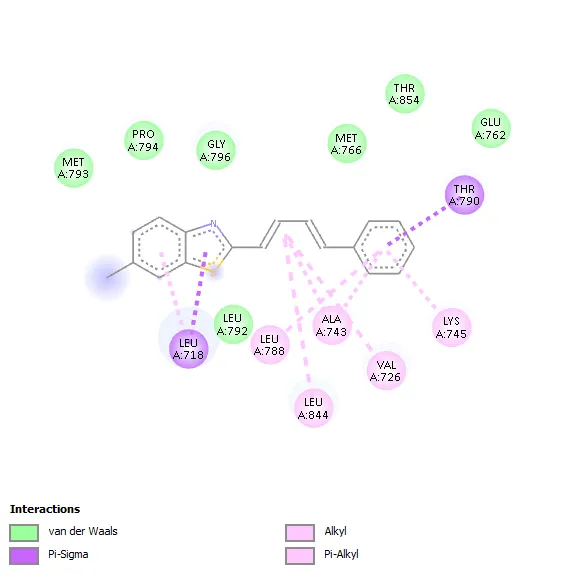
|
|
3e |
-7.6 |
Pro794 |
leu718, Thr790, Leu788, Ala743, Leu844, Val726, Lys745 |
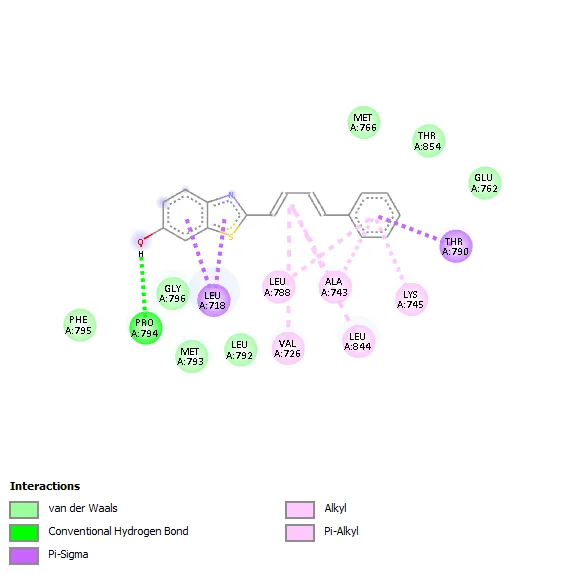
|
|
3f |
-7.6 |
- |
Leu718, Thr790, Val726, Leu788, Lys745, Ala743, Leu844 |

|
|
Std (Temoxifen) -7.4 |
- |
Leu844, Leu718, Ala743, Val726 |
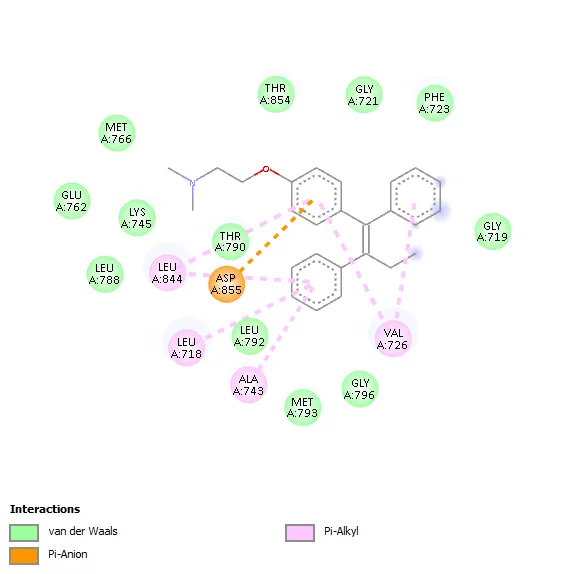
|
|
The molecular interaction analysis showed that hydrophobic interactions played a significant role in stabilizing the ligand–receptor complexes. The amino acid residues Leu718, Thr790, Ala743 Leu788, Leu844Val726, and Lys745 were commonly involved in ligand binding for most of the synthesized derivatives. These residues are considered important components of the ATP-binding pocket of EGFR kinase and contribute substantially to ligand stabilization through hydrophobic and van der Waals interactions.
Table: Binding Affinity of Synthesized compounds (3a-e) with target EGFR Kinase Domain (PDB ID: 2J6M)
|
Compound Code |
Substitution |
Binding Affinity (kcal/mol) |
|
3a |
3-NO₂ |
−7.6 |
|
3b |
4-OCH₃ |
−7.5 |
|
3c |
4-Cl |
−7.7 |
|
3d |
4-CH₃ |
−7.8 |
|
3e |
4-OH |
−7.6 |
|
3f |
3,4-Cl |
−7.6 |
|
Std. (Tamoxifen) |
— |
−7.4 |
Compound 3a showed a binding affinity of −7.6 kcal/mol and formed a hydrogen bond with Leu788 along with hydrophobic interactions involving Val726, Ala743, Leu844, and Leu718. Compounds 3b and 3c exhibited docking scores of −7.5 and −7.7 kcal/mol, respectively, with major hydrophobic interactions involving Leu718, Thr790, Ala743, Leu788, Leu844, Val726, and Lys745. Among all derivatives, compound 3d demonstrated the highest binding affinity (−7.8 kcal/mol) due to extensive hydrophobic interactions with key active-site residues. Compound 3e showed a docking score of −7.6 kcal/mol and formed a hydrogen bond with Pro794 along with several hydrophobic contacts. Similarly, compound 3f exhibited a binding affinity of −7.6 kcal/mol with interaction patterns comparable to the standard drug. The standard Tamoxifen showed a docking score of −7.4 kcal/mol with mainly hydrophobic interactions. Overall, several synthesized derivatives displayed better binding affinity than tamoxifen, indicating promising EGFR inhibitory potential.
The docking study suggested that the synthesized derivatives possess favorable interaction profiles within the EGFR kinase active site. The higher binding affinities and extensive hydrophobic interactions observed for compounds particularly 3c and 3d indicate their potential for further biological evaluation as prospective EGFR-targeted anticancer agents.
Molecular Docking Analysis of Synthesized Benzothiazole Derivatives Against Human Estrogen Receptor α (PDB ID: 3ERT)
The synthesized benzothiazole derivatives (3a–3f) were evaluated for their binding affinity and interaction pattern against the Human Estrogen Receptor Alpha ligand-binding domain using the crystal structure of Human Estrogen Receptor Alpha. The docking results were compared with the standard drug Tamoxifen to assess their potential as estrogen receptor-targeted anticancer agents.
Table: 2D Interaction and Binding Affinity of Benzothiazole Derivatives Against Human Estrogen Receptor Alpha Ligand (PDB ID: 3ERT)
|
Comp |
Binding Affinity (kcal/mol) |
H-Bond Interactions |
π–π / Hydrophobic Interactions |
2D Interaction |
|
3a |
-8.5 |
- |
Leu354, Trp383, Leu536, Ala350, Leu525, Leu346 |

|
|
3b |
-7.3 |
Thr347 |
Leu525, Lys529, Ala350, Leu391, Leu346, Leu387 |

|
|
3c |
-7.4 |
Thr347 |
Leu525, Leu387, Leu346, Leu391, Ala350, Met 343 |

|
|
3d |
-7.6 |
- |
Leu346, Ala350, Leu378, Leu391, Leu525, Met528 |

|
|
3e |
-7.7 |
Phe404 |
Leu391, Leu346, Leu387, Leu525, Leu384, Trp383, Ala350 |

|
|
3f |
-7.6 |
- |
Trp383, Ala350, Leu525, Met388, Phe404, Met421, Leu346 |

|
|
Std (Temoxifen) -8.6 |
- |
Ala350, Leu387, Leu346, Leu525, Met388, Met421 |
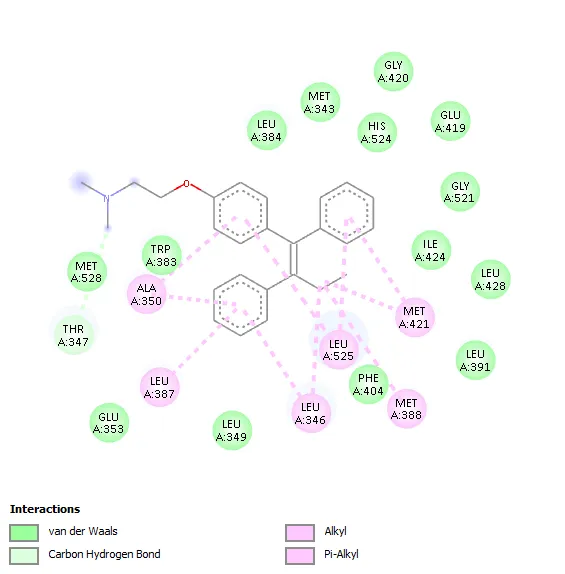
|
|
The molecular docking study against Human Estrogen Receptor Alpha revealed that all synthesized benzothiazole scaffolds shows favorable binding interactions within the active site of the receptor, with docking scores −7.3 to −8.5 kcal/mol. Among the tested compounds, derivative 3a showed the highest binding affinity (−8.5 kcal/mol), which was comparable to the standard drug Tamoxifen (−8.6 kcal/mol), indicating strong ligand–receptor interaction. Hydrophobic interactions played a major role in stabilization of the ligand–protein complexes, involving key amino acid residues such as Leu346, Ala350, Leu387, Leu391, Leu525, Trp383, Met388, and Phe404. Compound 3a interacted hydrophobically with Leu354, Trp383, Leu536, Ala350, Leu525, and Leu346. Compounds 3b and 3c formed hydrogen bonds with Thr347 along with multiple hydrophobic interactions, while compound 3e formed a hydrogen bond with Phe404 and showed extensive hydrophobic contacts. Compounds 3d and 3f mainly exhibited hydrophobic interactions with important active-site residues. The standard drug tamoxifen interacted predominantly through hydrophobic contacts involving Ala350, Leu387, Leu346, Leu525, Met388, and Met421.
Table: Binding Affinity of Synthesized compounds (3a-e) with target Human Estrogen Receptor Alpha Ligand (PDB ID: 3ERT)
|
Compound Code |
Substitution |
Binding Affinity (kcal/mol) |
|
3a |
3-NO₂ |
−8.5 |
|
3b |
4-OCH₃ |
−7.3 |
|
3c |
4-Cl |
−7.4 |
|
3d |
4-CH₃ |
−7.6 |
|
3e |
4-OH |
−7.7 |
|
3f |
3,4-Cl |
−7.6 |
|
Std. (Tamoxifen) |
— |
−8.6 |
The docking study indicated that the synthesized benzothiazole derivatives possess favorable binding characteristics against Human Estrogen Receptor Alpha. In particular, compound 3a exhibited excellent binding affinity and interaction profile, indicating its potential for further investigation as a prospective anticancer agent targeting estrogen receptor-positive breast cancer.
Synthesis Result
Benzothiazol-2amine Derivatives (2a–f)
Table: Physiochemical Properties of Synthesized derivatives (2a-f)
|
Com C |
R′ (Substituent) |
Mol. Wght |
Molecular For. |
% Yield (w/w) |
Color |
Rf |
m.p. (°C) |
|
2a |
3-NO2 |
195.20 |
C7H5N3O2S |
68% |
Dark yellow |
0.61 |
235-237 |
|
2b |
4-OCH3 |
180.23 |
C8H8N2OS |
59% |
Yellow |
0.72 |
210-212 |
|
2c |
4-Cl |
174.64 |
C7H5ClN2S |
62% |
Light Yellow |
0.58 |
232-234 |
|
2d |
4-CH3 |
164.23 |
C8H8N2S |
74% |
Pale Brown |
0.51 |
195-197 |
|
2e |
4-OH |
166.20 |
C7H6N2OS |
71% |
Yellow |
0.54 |
229-231 |
|
2f |
3,4-Cl |
219.08 |
C7H4Cl2N2S |
63% |
Light Brown |
0.63 |
240-242 |
Synthesis of N-(Substitutedbenzo[d]thiazol-2yl)-3-phenylprop-2-en-1imine Derivatives (3a–f)
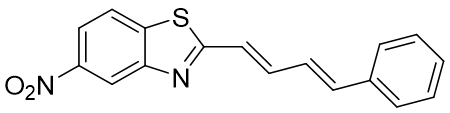
5nitro-2((1E,3E)-4phenylbuta-1,3-dien-1yl)benzothiazole (3a)
|
Mol. Form. |
C17H12N2O2S |
|
|
Mol. weight |
308.36 |
|
|
% yield (w/w) |
74 |
|
|
Color |
Dark Brown |
|
|
Melting point (°C) |
326-328 |
|
|
Solubility |
Soluble |
DMSO, chloroform, Acetone |
|
Partially soluble |
Methanol, ethanol |
|
|
Insoluble |
water |
|
|
Rf value |
0.51 |
|
TLC Solvent: Chloroform: Methanol (9:1 v/v)
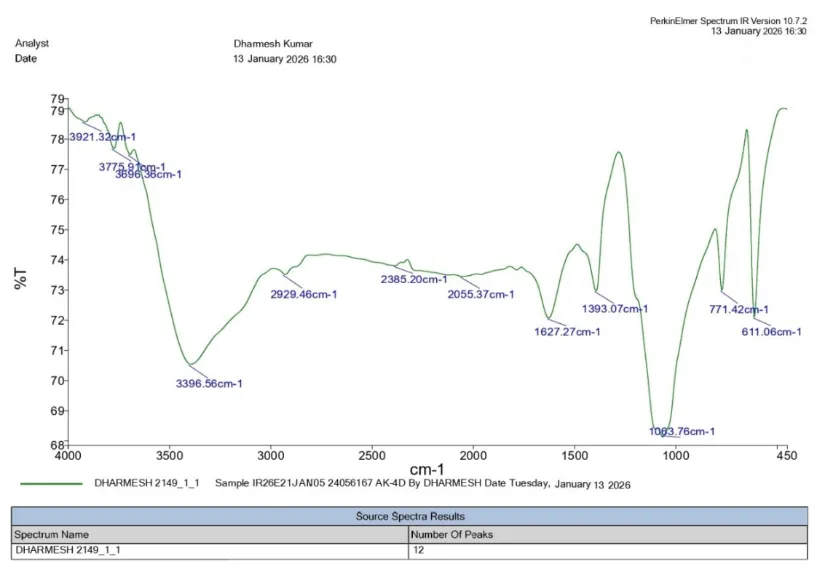
IR (KBr, cm⁻¹):
|
S. No. |
Functional Group |
Reference IR Range (cm⁻¹) |
Observed (cm⁻¹) |
|
1 |
Aromatic C=C |
1450–1600 |
1627.27 |
|
2 |
Aromatic C–H |
3000–3100 |
2929.46 |
|
3 |
Benzothiazole Ring (C=N) |
1600–1690 |
1627.27 |
|
4 |
Benzothiazole Ring (C–S) |
600–750 |
611.06 |
|
5 |
Aliphatic C=C |
1620–1680 |
1627.27 |
|
6 |
Nitro Group (C–NO₂) |
1340–1550 |
1393.07 |
¹H NMR (δ, ppm):
|

S. No. |
Proton Type |
Shift δ (ppm) |
No of H |
|
1 |
Ali C–H |
6.658–7.029 |
m, 4H, Aliphatic Hydrogen |
|
2 |
Ar–H |
7.081–7.668 |
m, 8H, Aromatic Hydrogen |
Mass Spectra:

|
S. No. |
Peak |
m/z Value |
Assignment |
|
1 |
Molecular Ion Peak |
308.31 |
[M⁺] |
|
2 |
Isotopic Molecular Ion Peak |
309.29 |
[M+1]⁺ |
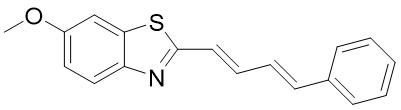
6methoxy-2-((1E,3E)-4phenylbuta-1,3dien-1-yl)benzothiazole (3b)
|
Mol. Form. |
C18H15NOS |
|
|
Mol. weight |
293.38 |
|
|
% yield (w/w) |
67 |
|
|
Color |
Dark Brown |
|
|
Melting point (°C) |
344-346 |
|
|
Solubility |
Soluble |
DMSO, chloroform, Acetone |
|
Partially soluble |
Methanol, ethanol |
|
|
Insoluble |
water |
|
|
Rf value |
0.56 |
|
IR (KBr, cm⁻¹):
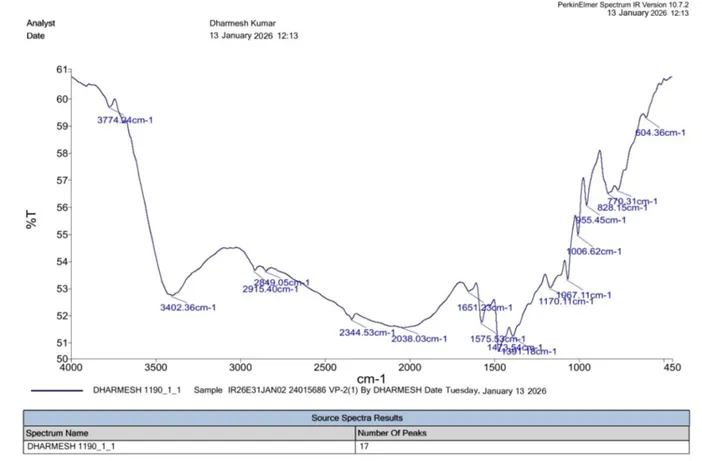
|
S. No. |
Functional Group |
Reference IR Range (cm⁻¹) |
Observed (cm⁻¹) |
|
1 |
Aromatic C=C |
1450–1600 |
1575.53 |
|
2 |
Aromatic C–H |
3000–3100 |
2915.40 |
|
3 |
Benzothiazole Ring (C=N) |
1600–1690 |
1651.23 |
|
4 |
Benzothiazole Ring (C–S) |
600–750 |
604.36 |
|
5 |
Aliphatic C=C |
1620–1680 |
1651.23 |
|
6 |
C–OH |
3200–3600 |
3402.36 |
¹H NMR (δ, ppm):

|
S. No. |
Proton Type |
Shift δ (ppm) |
No of H |
|
1 |
O–CH₃ |
3.826 |
s, 3H, Methoxy Proton (C–OCH₃) |
|
2 |
Ali C–H |
6.380–6.665 |
m, 4H, Aliphatic Hydrogen |
|
3 |
Ar–C–H |
7.490–8.240 |
m, 8H, Aromatic Hydrogen |
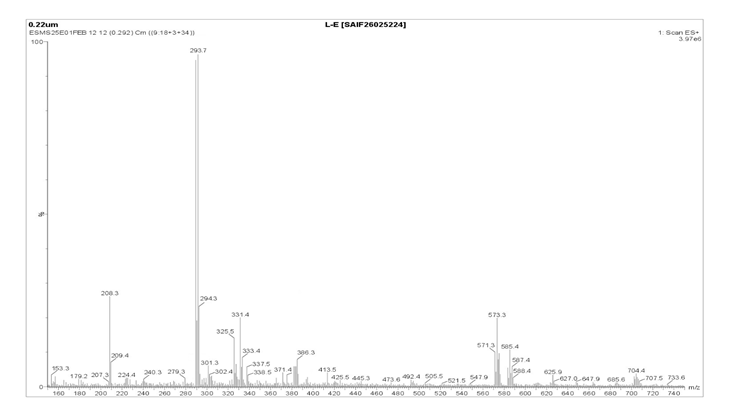
|
S. No. |
Peak |
m/z Value |
Assignment |
|
1 |
Molecular Ion Peak |
293.7 |
[M⁺] |
|
2 |
Isotopic Molecular Ion |
294.3 |
[M+1]⁺ |
6-chloro-2-((1E3E)-4phenylbuta-1,3dien-1yl)benzothiazole (3c)
|
Mol. Form. |
C17H12ClNS |
|
|
Mol. weight |
297.80 |
|
|
% yield (w/w) |
64 |
|
|
Color |
Dark Yellow |
|
|
Melting point (°C) |
224-226 |
|
|
Solubility |
Soluble |
DMSO, chloroform, Acetone |
|
Partially soluble |
Methanol, ethanol |
|
|
Insoluble |
water |
|
|
Rf value |
0.54 |
|
IR (KBr, cm⁻¹):
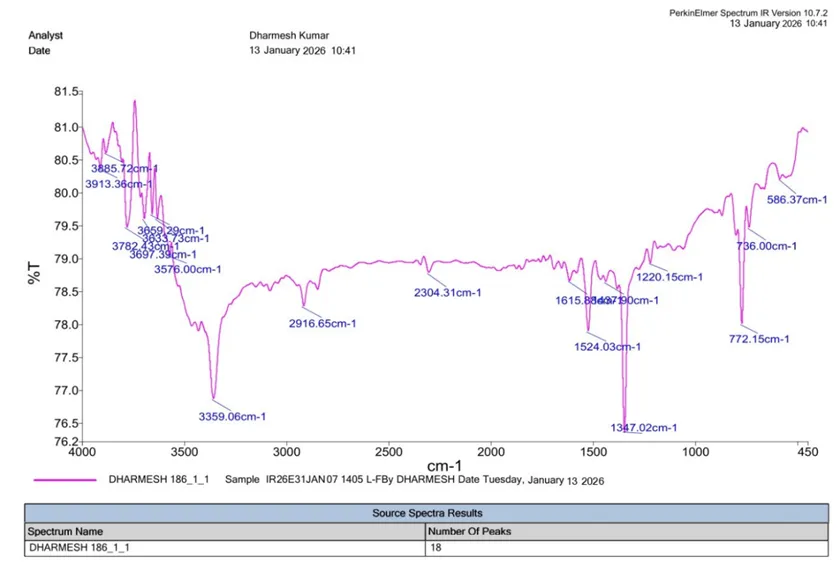
|
S. No. |
Functional Group |
Reference IR Range (cm⁻¹) |
Observed (cm⁻¹) |
|
1 |
Aromatic C=C |
1450–1600 |
1524.03 |
|
2 |
Aromatic C–H |
3000–3100 |
2916.65 |
|
3 |
Benzothiazole Ring (C=N) |
1600–1690 |
1615.88 |
|
4 |
Benzothiazole Ring (C–S) |
600–750 |
736.00 |
|
5 |
Aliphatic C=C |
1620–1680 |
1615.88 |
|
6 |
C–Cl |
600–800 |
772.15 |
¹H NMR (δ, ppm):
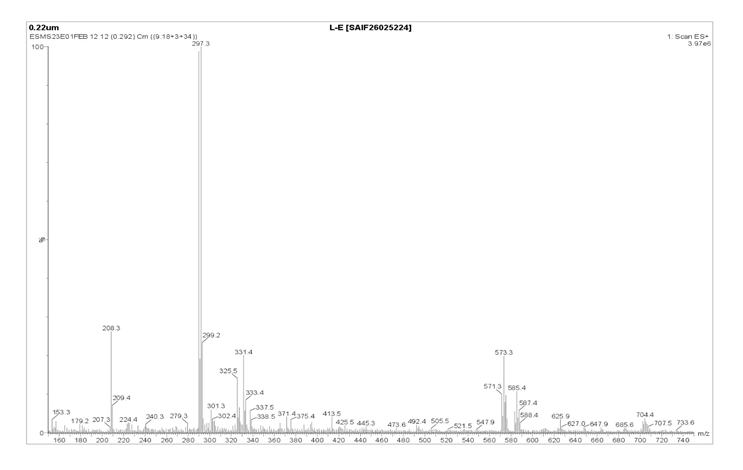
|
S. No. |
Proton Type |
Shift δ (ppm) |
No. of H |
||
|
1 |
Ali C–H |
6.775–7.217 |
m |
4H |
Aliphatic Hydrogen |
|
2 |
Ar–H |
7.321–7.821 |
m |
8H |
Aromatic Hydrogen |
|
S. No. |
Peak |
m/z Value |
Assignment |
|
1 |
Molecular Ion Peak |
297.3 |
[M⁺] |
|
2 |
Isotopic Molecular Ion Peak |
299.2 |
[M+2]⁺ |
6-methyl-2-((1E3E)-4phenylbuta-1,3-dien-1yl)benzothiazole (4d)
|
Mol. Form. |
C18H15NS |
|
|
Mol. weight |
277.39 |
|
|
% yield (w/w) |
76 |
|
|
Color |
Yellow |
|
|
Melting point (°C) |
304-306 |
|
|
Solubility |
Soluble |
DMSO, chloroform, Acetone |
|
Partially soluble |
Methanol, ethanol |
|
|
Insoluble |
water |
|
|
Rf value |
0.61 |
|
IR (KBr, cm⁻¹):
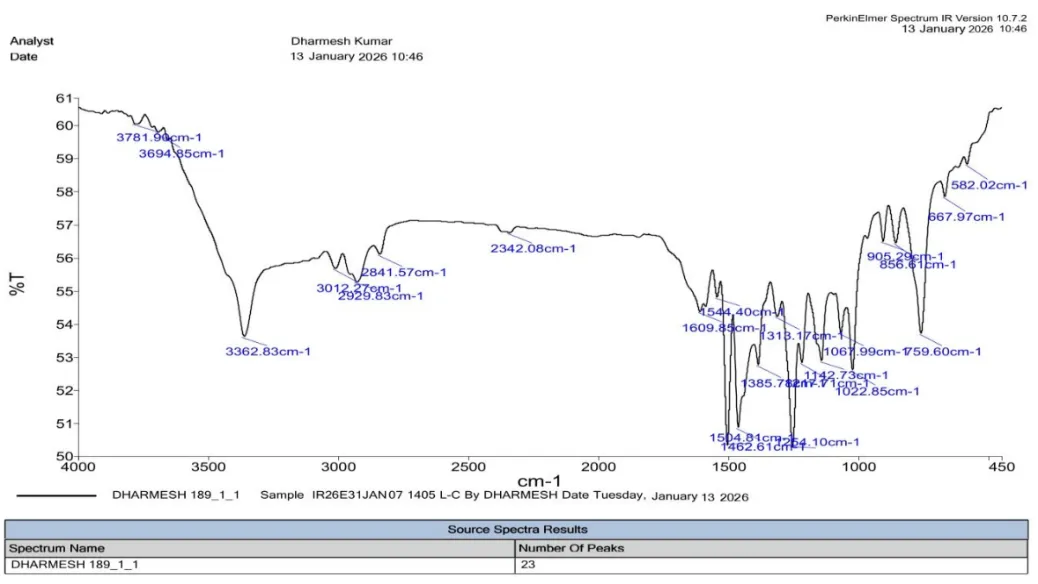
|
S. No. |
Functional Group |
Reference Range (cm⁻¹) |
Observed (cm⁻¹) |
|
1 |
Aromatic C=C |
1450–1600 |
1544.40 |
|
2 |
Aromatic C–H |
3000–3100 |
3012.27 |
|
3 |
Benzothiazole Ring (C=N) |
1600–1690 |
1609.85 |
|
4 |
Benzothiazole Ring (C–S) |
600–750 |
667.97 |
|
5 |
Aliphatic C=C |
1620–1680 |
1609.85 |
|
6 |
Aliphatic C–H |
2850–2960 |
2929.83 |
¹H NMR (δ, ppm):
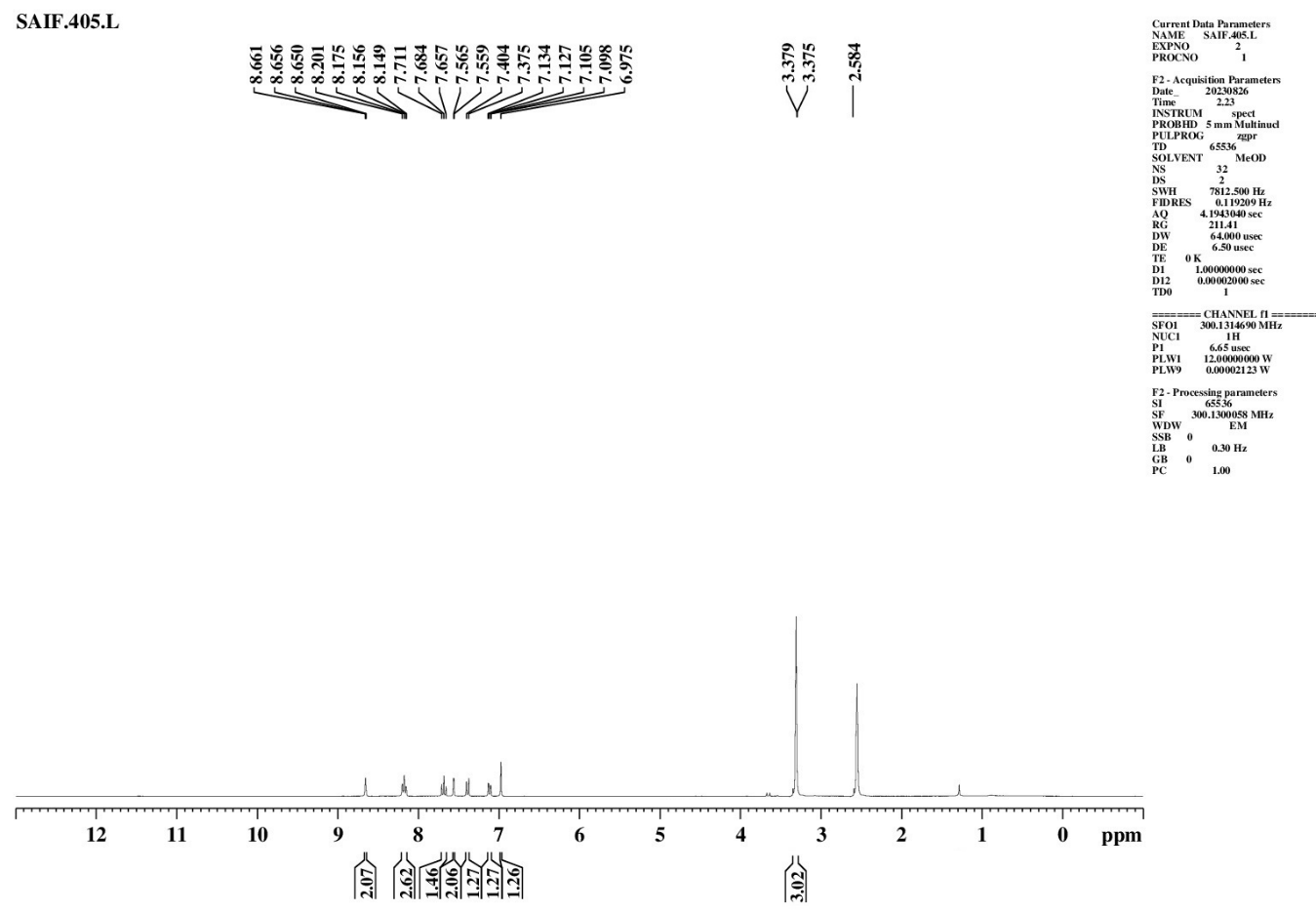
|
S. No. |
Proton Type |
Shift δ (ppm) |
Number of Protons |
||
|
1 |
Ali C–H |
3.379 |
m |
3H |
Aliphatic Hydrogen |
|
2 |
Ali C–H (HC=C) |
6.975–7.565 |
m |
4H |
Olefinic/Aliphatic Hydrogen |
|
3 |
Ar–H |
7.657–8.661 |
m |
8H |
Aromatic Hydrog |
2-((1E,3E)-4phenylbuta-1,3-dien-1-yl)benzothiazol-6ol (4e)
|
Mol. Form. |
C17H13NOS |
|
|
Mol. weight |
279.36 |
|
|
% yield (w/w) |
62 |
|
|
Color |
Yellow |
|
|
Melting point (°C) |
235-237 |
|
|
Solubility |
Soluble |
DMSO, chloroform, Acetone |
|
Partially soluble |
Methanol, ethanol |
|
|
Insoluble |
water |
|
|
Rf value |
0.68 |
|
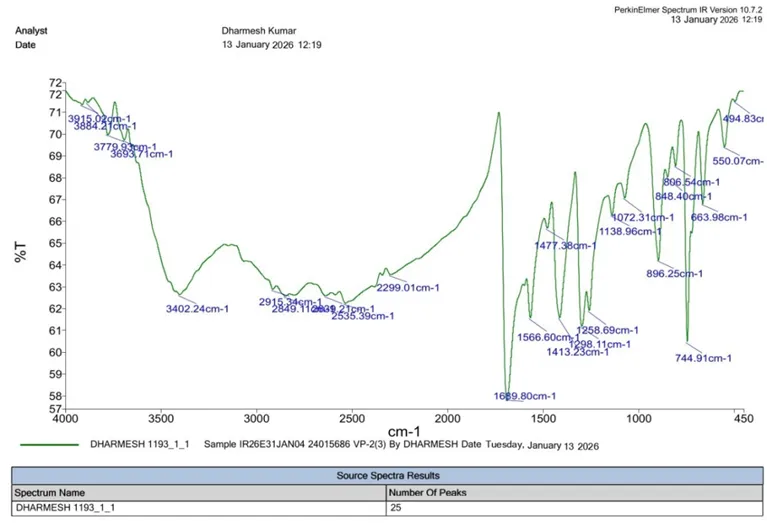
|
S. No. |
Functional Group |
Reference IR Range (cm⁻¹) |
Observed (cm⁻¹) |
|
1 |
Aromatic C=C |
1450–1600 |
1566.60 |
|
2 |
Aromatic C–H |
3000–3100 |
2915.34 |
|
3 |
Benzothiazole Ring (C=N) |
1600–1690 |
1689.80 |
|
4 |
Benzothiazole Ring (C–S) |
600–750 |
744.91 |
|
5 |
Aliphatic C=C |
1620–1680 |
1689.80 |
|
6 |
C–OH |
3200–3600 |
3402.24 |
¹H NMR (δ, ppm):
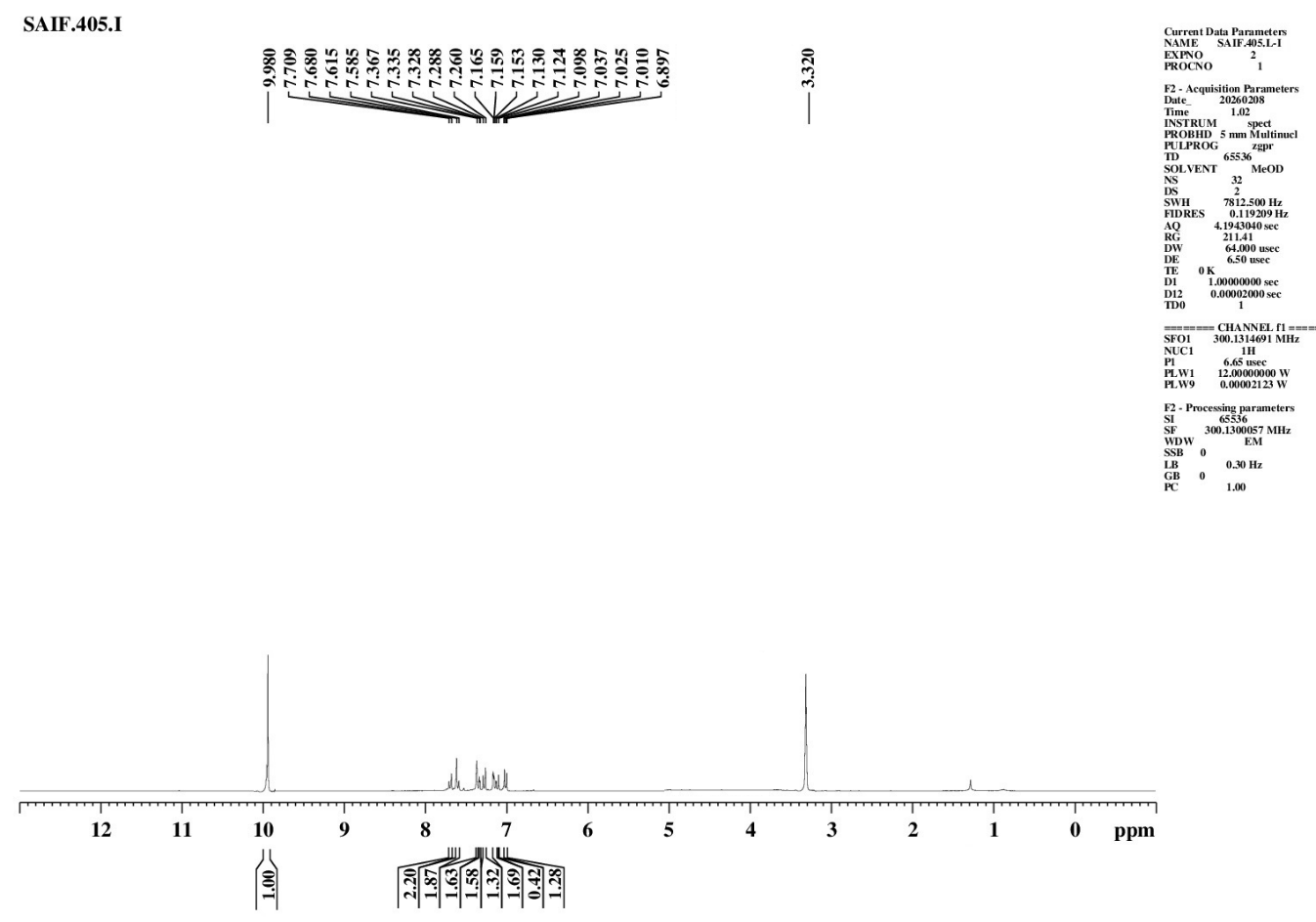
|
S. No. |
Proton Type |
Shift δ (ppm) |
Number of Protons |
||
|
1 |
Ali C–H |
6.897–7.153 |
m |
4H |
Aliphatic Hydrogen |
|
2 |
Ar C–H |
7.159–7.709 |
m |
8H |
Aromatic Hydrogen |
|
3 |
Ar O–H |
9.980 |
s |
1H |
Hydroxyl Proton |

|
S. No. |
Peak |
m/z Value |
Assignment |
|
1 |
Molecular Ion Peak |
279.9 |
[M⁺] |
|
2 |
Isotopic Molecular Ion Peak |
280.30 |
[M+1]⁺ |
5,6-dichloro-2((1E3E)-4phenylbuta-1,3-dien-1yl)benzothiazole (3f)
|
Mol. Form. |
C18H15NOS |
|
|
Mol. weight |
332.08 |
|
|
% yield (w/w) |
67 |
|
|
Color |
Dark Brown |
|
|
Melting point (°C) |
344-346 |
|
|
Solubility |
Soluble |
DMSO, chloroform, Acetone |
|
Partially soluble |
Methanol, ethanol |
|
|
Insoluble |
water |
|
|
Rf value |
0.72 |
|
IR (KBr, cm⁻¹):
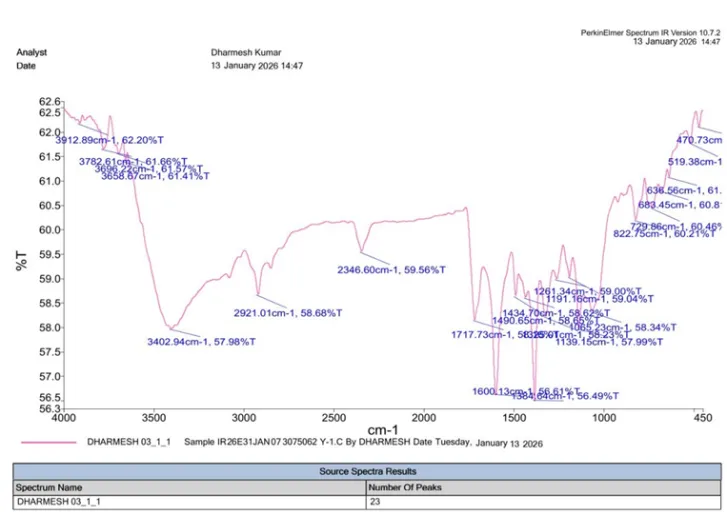
|
No. |
Functional Group |
Reference IR Range (cm⁻¹) |
Observed (cm⁻¹) |
|
1 |
Aromatic C=C |
1450–1600 |
1490.65 |
|
2 |
Aromatic C–H |
3000–3100 |
2921.01 |
|
3 |
Benzothiazole Ring (C=N) |
1600–1690 |
1600.13 |
|
4 |
Benzothiazole Ring (C–S) |
600–750 |
683.45 |
|
5 |
Aliphatic C=C |
1620–1680 |
1717.73 |
|
6 |
C–Cl |
600–800 |
729.86 |
Mass Spectra:
|
S. No. |
Peak |
m/z Value |
Assignment |
|
1 |
Molecular Ion Peak |
332.44 |
[M⁺] |
|
2 |
Isotopic Molecular Ion Peak |
333.58 |
[M+1]⁺ |
Antioxidant Activity by DPPH Assay
The DPPH free radical scavenging assay was used to assess the antioxidant activity of the synthesized benzothiazole scaffolds (3a–3f), and the results were compared with those of the conventional antioxidant Ascorbic Acid. The assay was carried out at various doses between 20 and 100 µg/mL, and the % inhibition of DPPH radicals was measured using UV at 517 nm. All synthesized compounds demonstrated concentration-dependent free radical scavenging activity. An increase in percentage inhibition was observed with increasing concentration of the test compounds, indicating effective antioxidant potential of the synthesized benzothiazole derivatives. The reduction in absorbance of the DPPH solution from deep violet to pale yellow confirmed the hydrogen-donating and radical-neutralizing ability of the compounds.
Table: Absorption of Synthesized compounds (3a-f)
|
Conc. (µg/mL) |
Absorption |
|||||||
|
Control Absorbance (Ac) |
3a (As) |
3b (As) |
3c (As) |
3d (As) |
3e (As) |
3f (As) |
Std. Ascorbic Acid (As) |
|
|
20 |
0.850 |
0.575 |
0.641 |
0.665 |
0.616 |
0.521 |
0.647 |
0.438 |
|
40 |
0.850 |
0.469 |
0.541 |
0.569 |
0.515 |
0.401 |
0.547 |
0.309 |
|
60 |
0.850 |
0.366 |
0.436 |
0.488 |
0.415 |
0.281 |
0.452 |
0.188 |
|
80 |
0.850 |
0.276 |
0.359 |
0.394 |
0.322 |
0.175 |
0.377 |
0.088 |
|
100 |
0.850 |
0.211 |
0.290 |
0.328 |
0.252 |
0.100 |
0.309 |
0.027 |
Among the synthesized benzothiazole derivatives, compound 3e exhibited the highest antioxidant activity, showing 38.7–88.2% inhibition over the concentration range of 20–100 µg/mL, which may be attributed to the presence of the hydroxyl substituent that enhances hydrogen donation and free radical stabilization. Compound 3a also demonstrated considerable antioxidant potential with 75.2% inhibition at 100 µg/mL, possibly due to the nitro substituent and conjugated benzothiazole Schiff base system. Compound 3d showed moderate activity, while compounds 3b and 3f exhibited intermediate free radical scavenging effects. In contrast, compound 3c displayed comparatively lower antioxidant activity (61.4% inhibition at 100 µg/mL), which may be associated with the electron-withdrawing nature of the chloro substituent. The standard antioxidant Ascorbic Acid showed the highest activity with 96.8% inhibition at 100 µg/mL. Overall, the synthesized derivatives demonstrated appreciable antioxidant potential, with activity significantly influenced by the nature of substituents attached to the benzothiazole nucleus.
The results suggested that antioxidant activity of the synthesized compounds was significantly influenced by the nature and position of substituents attached to the benzothiazole nucleus. Electron-donating groups such as hydroxyl substituents enhanced the antioxidant potential, whereas electron-withdrawing groups exhibited comparatively lower activity. The presence of conjugated aromatic systems and Schiff base linkage may also contribute toward stabilization of free radicals through electron delocalization mechanisms.
Table: Percentage Inhibition of Synthesized compounds (3a-f)
|
Concentration (µg/mL) |
(% Inhibition ± SD) |
||||||
|
3a |
3b |
3c |
3d |
3e |
3f |
Std. Ascorbic Acid |
|
|
20 |
32.4 ± 0.8 |
24.6 ± 0.6 |
21.8 ± 0.5 |
27.5 ± 0.7 |
38.7 ± 0.9 |
23.9 ± 0.6 |
48.5 ± 1.1 |
|
40 |
44.8 ± 1.0 |
36.3 ± 0.8 |
33.1 ± 0.7 |
39.4 ± 0.9 |
52.8 ± 1.2 |
35.7 ± 0.8 |
63.7 ± 1.4 |
|
60 |
56.9 ± 1.2 |
48.7 ± 1.1 |
42.6 ± 0.9 |
51.2 ± 1.1 |
66.9 ± 1.5 |
46.8 ± 1.0 |
77.9 ± 1.7 |
|
80 |
67.5 ± 1.4 |
57.8 ± 1.3 |
53.7 ± 1.1 |
62.1 ± 1.3 |
79.4 ± 1.8 |
55.6 ± 1.2 |
89.6 ± 1.9 |
|
100 |
75.2 ± 1.6 |
65.9 ± 1.4 |
61.4 ± 1.3 |
70.3 ± 1.5 |
88.2 ± 2.0 |
63.7 ± 1.4 |
96.8 ± 2.1 |
Based on the obtained results, compound 3e emerged as the most potent antioxidant derivative among the synthesized compounds and may serve as a promising candidate for further pharmacological and biological investigations.
CONCLUSION
The findings of the present investigation suggest that benzothiazole-based Schiff base derivatives possess considerable potential as multifunctional molecules exhibiting both anticancer and antioxidant properties. The combination of favorable molecular docking interactions and appreciable antioxidant activity highlights the importance of benzothiazole scaffolds in medicinal chemistry and supports further pharmacological and biological exploration of these derivatives for development of safer and more effective therapeutic agents against breast cancer and oxidative stress–related disorders.
REFERENCES

Anjali Gupta, Sarita, Ankit Kumar, Synthesis, Characterization, In-Silico Anticancer Evaluation & In-Vivo Antioxidant Evaluation of Novel Benzothiazole Derivatives, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 6381-6408, https://doi.org/10.5281/zenodo.20845001
 10.5281/zenodo.20845001
10.5281/zenodo.20845001