
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Dr. R. G. Bhoyar Institute of Pharmaceutical Education & Research, Wardha, Maharashtra
Computer-Aided Drug Discovery (CADD) utilizes theoretical and computational methods like computational chemistry and molecular modeling to enhance drug development. Key techniques include 3D-QSAR pharmacophore modeling, which links molecular structure to biological activity, and Virtual Screening (VS) for assessing large compound libraries. Molecular Dynamics (MD) simulations improve drug design accuracy by providing insights into molecular interactions. The Drug Bank database offers extensive drug and target information, while other databases like HMDB and FooDB provide additional data. AutoDock, a simulation program for molecular modeling, is widely used for protein-ligand docking, with its successor AutoDock Vina offering enhanced capabilities, both maintained by the Center for Computational Structural Biology at Scripps Research. AutoDock has been crucial in developing clinically approved HIV-1 integrase inhibitors.
1. Computer- Aided Drug Discovery (CADD)
A wide range of theoretical and computational methods used in a contemporary drug discovery are included in Computer Aided Drug Design (CADD). The development of medication that are currently in clinical use or undergoing clinical trials has benefited greatly from the application of CADD techniques. These techniques have developed alongside the experimental strategies employed in the drug design.
A contemporary computational method for finding and developing a possible lead in the drug discovery process is called CADD. Computational chemistry, molecular modeling, molecular design, and rational drug design are all included in computer aided drug design. To optimize identified leads, CADD is utilized. Both academia and the pharmaceutical industry are beginning to recognize and value CADD techniques.
2. 3D QSAR pharmacophore modeling
3D-QSAR is a powerful technique that establishes a quantitative connection between molecules biological activities and their threedimensional structural characteristics. By analyzing how variations in molecular shape, charge distribution, and hydrophobicity affect the binding affinity of compounds to a given biological target, 3D-QSAR provides predictive models that can guide the design of more potent molecules. When combined with pharmacophore modeling, which identifies key molecular features required for target binding, this approach becomes a potent tool in virtual drug design. Pharmacophore models focus on the spatial configuration of functional groups necessary for activity, offering a framework for creating novel molecules with improved biological efficacy.
3. Virtual screening
Virtual Screening (VS) makes it possible to quickly screen large compound libraries in silico; it has completely changed the early phases of drug discovery. The technique uses computational algorithms to dock molecules into a biological targets three-dimensional structure, assessing their binding affinity and forecasting their potential as lead compounds. Even from large compound libraries, molecules with advantageous binding properties can be found using ligand-based and structure-based virtual screening technique, or combination of these. This simplifies and lowers the cost of the discovery process by significantly reducing the number of compound that must be synthesized and test experimentally.
4. Molecular dynamic simulation
Molecular Dynamics (MD) Simulations offer a deeper understanding of molecular behavior over time, allowing researchers to investigate not only the binding affinity but also the dynamic interactions between the drug and its target, enhancing the accuracy of drug design. In addition to offering a deeper, more nuanced understanding of molecular interaction, this all –encompassing approach can drastically cut the type and expense associated with conventional drug discovery techniques, enabling the logical design of medication with increased specificity and fewer side effects.
Software:
1. Drug bank:
The University of Alberta and the Metabolomics cheminformatics resource created and maintains the extensive, publicly available drug bank database, which combines detailed drug (i.e., chemical, pharmacological and innovation center located in Alberta, Canada) and drug target information. As both pharmaceutical and bioinformatics data with thorough drug target (i.e. sequence, structure, and pathway) information.The public can access the drug bank online website for free. However, license is needed for any use or repurposing of drug bank online content or the underlying drug bank data, in whole or in part, and for any purpose. While all other users need a paid license, academic users can apply for a free license for specific use cases. Four more databases, including a general suite Metabolomic/ cheminformatics database include HMDB, T3DB, SMPDB, and FooDB. Equivalent data on over 3100 common toxins and 40,000 human metabolic pathway and disease pathway can be found in SMPDB; and approximately 28,000 food components and food additives can be found in Food.
2. Autodock:
Autodock is a simulation program for molecular modeling. It works particularly well for protein-ligand docking. The GNU General Public License governs the availability of AutoDock 4. One of the docking software programs that researchers use the most frequently is AutoDock.
The accuracy and performance of AutoDock Vina, its replacement, have been greatly enhanced. The Apache license makes it accessible. Currently, Scripps Research- more especially, the Center for Computational Structural Biology (CCSB), headed by Dr. Arthur J. Olson-maintains both AutoDock and Vina. AutoDock is a popular tool that helped Merck and Co. develop the first HIV-1 integrase inhibitor that was clinically approved.
2. AIM AND OBJECTIVES
AIM
3D-QSAR-Based Pharmacophore Modeling, Virtual Screening, and Molecular Dynamics Simulations.
OBJECTIVE
1. To carry out comprehensive literature survey and selection of drug.
2. To make the drug discovery process cost efficient.
3. To reduce time required for drug development.
4. Finding a possible lead compound to increase the success rates of drug discovery projects.
5. Estimating lead compounds’ absorption, distribution, metabolism, and excretion (ADME) characteristics to guarantee their efficacy and safety.
6. Enhance the lead compounds’ affinity for the target protein.
3. PLAN OF WORK
The plan of work was carried out in the following systemic manner. An extensive literature survey carried out for the selection various computer -based method. According to literature commercially free software/webserver used in the work.
Literature Review
↓
TARGET Selection DRUG Selection
↓
Prediction of biological activities [PASS ONLINE]
↓
Identification of published activities
↓
Selection of new Activities for further studies
↓
[DRUG BANK]
[WAY – 2 – DRUG]
↓
Molecular Docking
[AUTODOCK TOOLS]
↓
Visualization of molecular docking result
[AUTODOCK TOOLS] [BIOVIA DISCOVERY STUDIO
4. DRUG PROFILE
1. Sample Drug: Seladelpar
Table no. 1: Drug profile – Seladelpar
|
Structure |

|
|
Synonym |
Livdelzi and Lyvdelzi |
|
Chemical Formula |
C21H23F3O5S |
|
Molecular Weight |
444.47 |
|
Indication |
Adults with primary biliary cholangitis (PBC) can be treated with Seladelpar, which is marketed under the brand name Livdelzi. Depending on how the patient responded to ursodeoxycholic acid (UDCA) in the past, it can be used either alone or in conjunction with UDCA. The particular indications that have been approved are: Adults with PBC who have not responded well to UDCA may benefit from combination therapy. Adults with PBC who cannot tolerate UDCA are treated with monotherapy. |
|
Pharmacodynamics |
Seladelpar works to decrease the levels of total bile acids and reduce bile acid synthesis in patients with PBC.1,4 Alkaline phosphatase (ALP) levels may rise in hepatobiliary disorders, such as PBC, due to an increase in bile acid concentration. 3 Compound to the placebo group, patient with PBC receiving 10 mg of Seladelpar once daily showed a greater decrease in mean ALP from baseline as early as one month after treatment, and the lower ALP was typically maintained through month 12. A dose – dependent decrease mean ALP was noted in another study where patients with PBC were given 2,5, or 10g of Seladelpar once daily. |
|
Associated Condition |
Seladelpar is associated with the liver disease Primary Biliary Cholangitis (PBC), for which it is approved as a treatment |
2. Standard Drug: Indomethacin
Table no. 2: Drug Profile - Indomethacin
|
Structure |
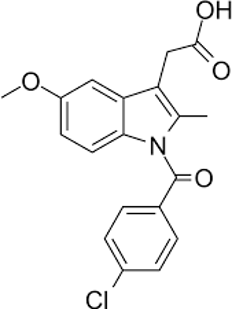
|
|
Synonym |
Indomethacin, Indomethacina, Indomethacine |
|
Chemical formula |
C19H16ClNO4 |
|
Molecular weight |
357.788 |
|
Indication |
For the treatment of moderate to severe rheumatoid arthritis symptoms, including acute flare-ups of the chronic condition, oral indomethacin is recommended. |
|
Associated conditions |
Indomethacin, a nonsteroidal anti-inflammatory drug (NSAID),is used to treat pain, swelling, and stiffness that come with a number of condition. |
|
Pharmacodynamics |
Indomethacin is and NSAID that works by stopping the production of substances that cause pain, fever, and inflammation. It has pain-reliving and fever-reducing effects. Its therapeutic effect does not entail stimulation of the pituitary-adrenal axis. Indomethacin primarily works by suppressing inflammation in rheumatoid arthritis by providing relief of pain as well as reducing fever, swelling, and tenderness. |
3. Standard Drug: Elafibranor
Table No. 3: Drug Profile: Elafibranor
|
Structure |
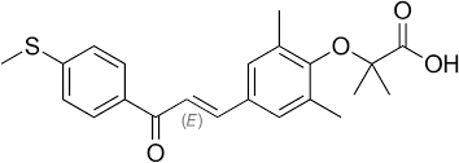
|
|
Synonym |
ELAFIBRANOR |
|
Chemical formula |
C22H24O4S |
|
Molecular weight |
384.49 |
|
Indication |
Elafibranor is indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have had an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA. |
|
Associated Conditions |
Elafibranor (brand name lqirvo) is a prescription medication primarily approve for the treatment of primary biliary cholangitis (PBC). |
|
Pharmacodynamics |
Elafibranor prevents the production of bile acids. Additionally, it has been demonstrated to enhance lipid metabolism, glucose homeostasis, and insulin sensitivity. One Elafibranor decreased the average level of alkaline phosphatase (ALP) in PBC patients. Both Elafibranor and GFT1007 activated PPARalpha, according to an in vitro PPAR functional assay (EC50=46 Nm and 14 Nm, respectively, relative to reference agonists). |
5. EXPERIMENTAL WORK
1. PASS ONLINE (Prediction of Activity Spectra for Substances):
Finding new targets (mechanisms) can be effectively accomplished with the PASS (prediction of Activity Spectra for substances) software product, which predicts over 300 pharmacological effects and biochemical mechanism based on a substance’s structural formula.
2. Drug bank database:
Go to https://go drugbank.com/ and search seladelpar to find the drug profile and SMILES (Simplified Molecular Input Line Entry System) file of seladelpar for additional research, as illustrated stepwise in figs. 1 and 2.
Figs: 1
Figs: 2
3. Way2Drug:
SMILES file is used to get predictions of activities of Seladelpar as given in figures Fig 3 and Fig 4. (https://way2drug.com/passonline/
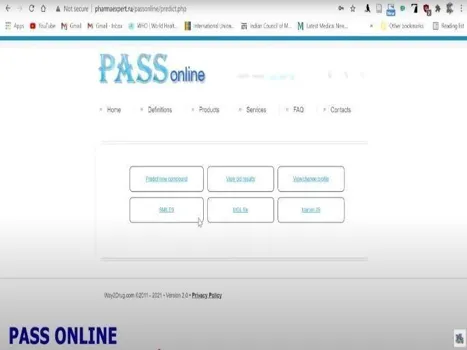
Fig: 3 Fig: 4
Docking Method
1. PDB (Protein Data Bank)
Search target on RCSB-PDB website to obtain their PDB file of target. Fig 5 and Fig 6. (https://www.rosh.org/structure/Zial)
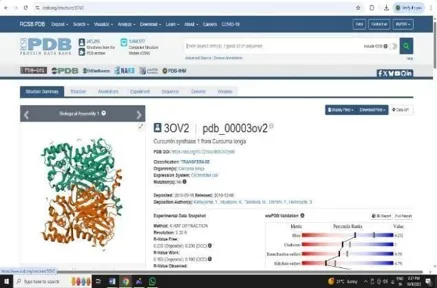
Fig: 5 Fig: 6
2. Drug Bank
Visit the drug bank website to download the PDB file of the sample drug needed for the docking process. (https://go.drugbank.com)
3. Making of Docking System
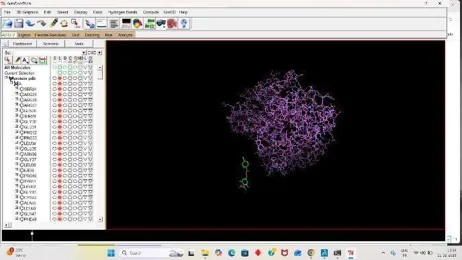
Open the Autodock tools software for further docking process, firstly make a folder in the C-Drive of the computer for the docking process, Then paste the PDB files of the target and the drug in that folder. We have to make a system for our docking process, so we have to copy some important files in the folder
i] Both autodock4.exe and autogrid4.exe
ii] AD4_parameters.dat and AD4.1_bound.dat
iii] vina.exe, vina_license.rtf and vina_split.exe
After successful copying of our important data in the folder we have to copy the address of our folder and paste it the autodock startup directory (for only showing the useful files).
Fig: 7
1. Protein Preparation
Under the file section click on Read molecule open our Receptor PDB, Clean the target PDB by removing its water from the edit section, Then add hydrogens (Polar Only), Then Add Charges from the edit section again firstly add kollmann charges, Then compute gasteiger charges.
After adding charges under the edit section, Click on Atoms and Assign AD4 type, Then save the receptor file in PDBQT format.
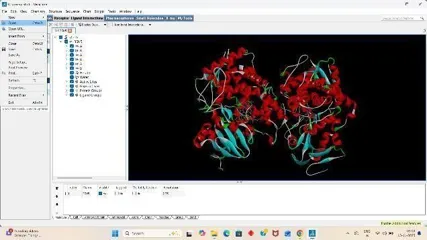
Fig: 8 Fig: 9

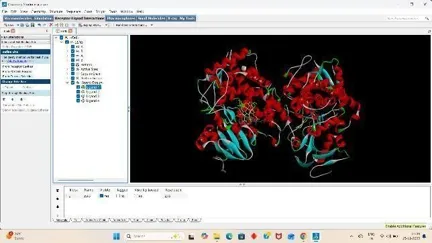
Fig: 10 Fig: 11
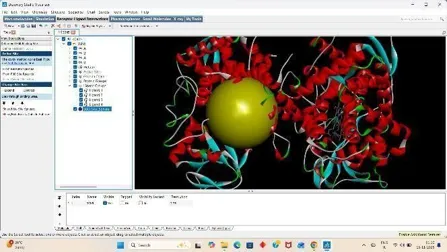
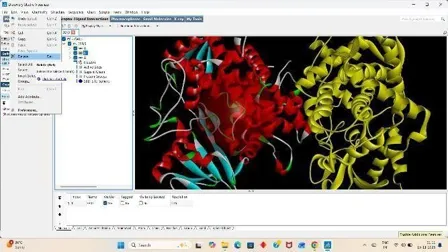
Fig: 12 Fig: 13
2. Ligand preparation
Additionally, we must get our ligand ready for the docking process. To do this, select the Ligand section, torsion tree, and finally select choose root. Next click ligand once more, select torsions (OK) under torsion tree, and then click output to save the prepared ligand file in pdbqt format. Fig 14 and Fig 21.
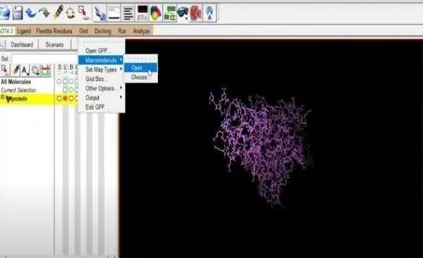

Fig: 14 Fig: 15
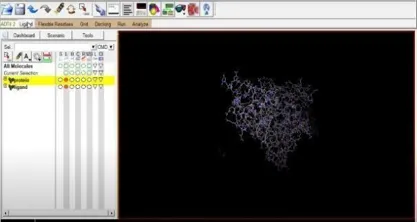
Fig: 16 Fig: 17
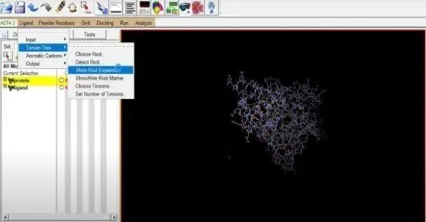

Fig: 18 Fig:19

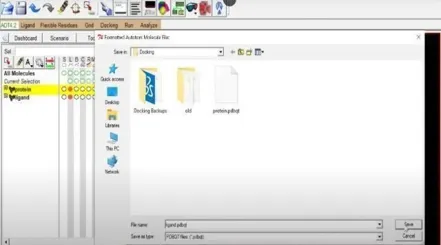
Fig: 20 Fig: 21
3. Grid Preparation
For Docking we have to prepare our ligand and receptor by applying grid, So click on grid, Click on macromolecule, Then choose and select our protein then a box appear click OK, and replace your receptor pdbqt with new receptor, pdbqt file. (22)
Then click on grid, under it click on Set map types and choose the ligand, Then for final docking process add grid box by on grid box and add grid box to your receptor and save the grid dimension file and then click on file and close saving current. (28)
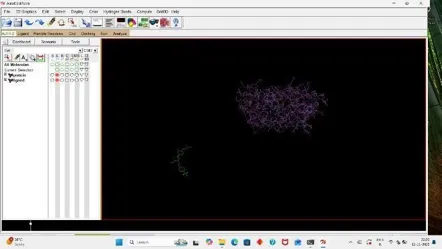
Fig: 22 Fig: 23
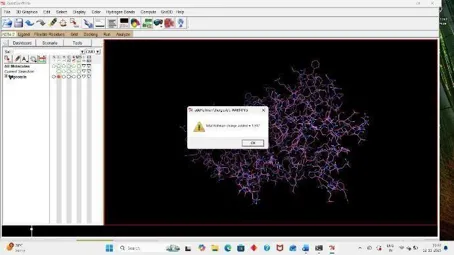
Fig: 24 Fig: 25

Fig: 26 Fig: 27
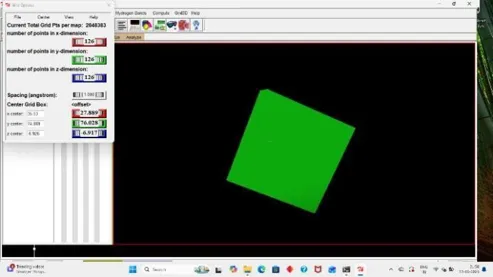
Fig: 28
4. Command process
For getting the docking result make a config. File in the same folder as in Fig 29 and add the x,y,z axis coordinates from the grid dimension file then save it as preferred. For final docking open command prompt from startup window and give command.
A log file is generated in the our docking folder that is our result.
Fig: 29 Fig: 30
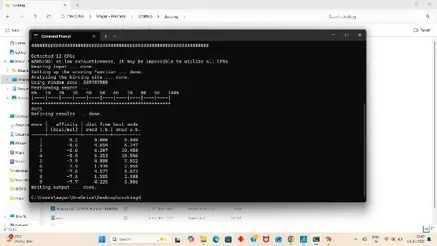
Fig: 31 Fig: 32
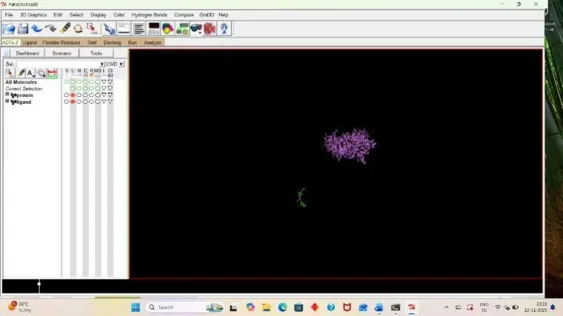
5. Visualization
For analyzing the result, open autodock software and click on Analyze, Then under docking click on open autodock vina results and select your log file that is out result. (33)
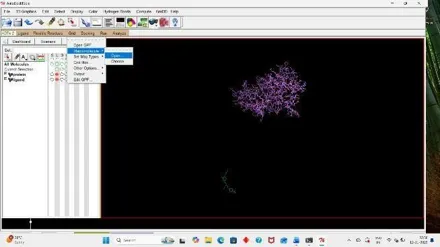
Fig: 33 Fig: 34
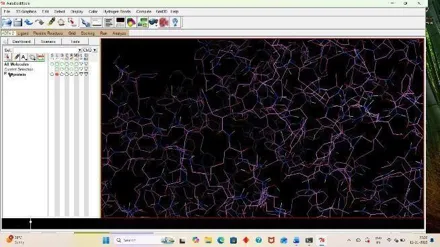
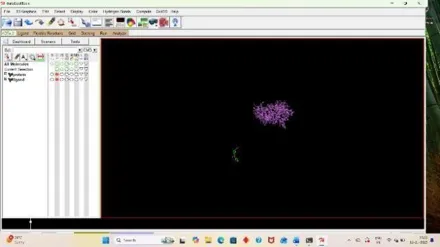
Fig: 35 Fig: 36
6. RESULTS AND DISCUSSION
1. OUTCOME
2. ACTIVITY THAT FIGHTS INFLAMMATION
|
SR. NO |
PARAMETERS |
DOCKING SCORE |
RESULT |
|
1] |
Sample: 1]Seladelpar + Target (PPAR◻) Amino acid residues: SER24, ARG25, ARG26, GLY37, LEU38 |
-9.1 |
|
|
2] |
Standard: 2] Indomethacin + Target (PPAR◻) Amino acid residues: LEU211, LYS212, PHE214, ILE218, TYR219 |
-9.4 |
|
3. ANTI- FIBROTIC ACTIVITY
|
SR NO. |
PARAMETERS |
DOCKING SCORE |
RESULT |
|
1] |
Sample drug: 1] Seladepar + Target (CYP7A1) Amino acid residue: HIS25,GLU36, ALA19, LEV27 |
-8.9 |
|
|
2] |
Standard drug 2] Elafibranor + Target (CYP7A1) Amino acid residue: SER21,MET22, THR22, ILE21, TYR22 |
-8.7 |
|
7. DISCUSSION
In the present work all docking work is done with the Autodock tools and ligand & receptors prepared according to the method & material. The receptor is docked individually with the standard (Indomethacin and Elafibranor) and sample (seladepar) with the PPAR and CYP7A1 respectively. The receptor Peroxisome proliferator- activated receptor delta (PPAR) use for sample (seladelpar) and standard (Indomethacin) interaction score (docking score) was found ∆G =-9.1, -9.3 respectively it shows that seladelpar may be have activity toward PPAR◻ than indomethacin that’s why it may use as the anti-inflammatory activity. The receptor cholesterol 7 alpha-hydroxylase (CYP7A1) use for sample (seladelpar) and standard (Elafibranor) interaction score (docking score) was found ∆G = -8.9, -8.7 respectively It shows that seladelpar has binding affinity to the receptor, it concludes that the seladelpar shows better activity as antifibrotic activity.
8. CONCLUSION
After performing docking of standard (Indomethacin and Elafibranor) and sample (seladelpar) with PPAR◻ and CYP7A1 the score was found to be ∆G= -9.1, -9.3 and -8.9, -8.7 respectively. This score explains that seladelpar shows activity on PPAR-◻ (Peroxi-some proliferatar activated receptor delta) and cholesterol 7 alpha-hydroxylase (CYP7A1) which shows that seladelpar may show anti- inflammatory and anti- fibrotic action due to this we can use seladepar as anti-fibrotic and anti- inflammatory agent which also use in liver fibrosis.
ACKNOWLEDGEMENT
This project was conducted in Dr. R. G. Bhoyar Institute of Pharmaceutical Education &Research, Wardha, Maharashtra.
REFERENCES





Mayur Ambatkar, Suhani Korde, Kaveri Rathod, Nikita Lidabe, Nitin Indurwade, 3D-QSAR-Based Pharmacophore Modeling, Virtual Screening, and Molecular Dynamics Simulations of Seladelpar, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 7385-7397. https://doi.org/10.5281/zenodo.21044435
 10.5281/zenodo.21044435
10.5281/zenodo.21044435