Poor aqueous solubility is a major limitation affecting the bioavailability of many newly developed pharmaceutical compounds. A significant proportion of drugs classified under the Biopharmaceutics Classification System (BCS) exhibit low solubility, which results in poor dissolution rate and limited therapeutic efficacy. Various formulation strategies have been explored to overcome this challenge, among which solid dispersion has emerged as an effective and widely investigated approach for improving drug solubility and dissolution behavior. Solid dispersion involves dispersing a poorly soluble drug within a hydrophilic carrier matrix, which enhances wettability, reduces particle size, and may convert the drug from crystalline to amorphous form, thereby increasing dissolution rate and bioavailability. Over the years, several preparation techniques such as fusion method, solvent evaporation, spray drying, hot-melt extrusion, and lyophilization have been developed to produce solid dispersions with improved physicochemical properties. In addition, different generations of solid dispersion systems using polymers, surfactants, and carrier combinations have been investigated to enhance stability and drug release characteristics. Despite their advantages, challenges such as physical instability, recrystallization, and scale-up limitations remain important considerations during formulation development. This review highlights the fundamental principles of solid dispersion technology, classification of solid dispersions, commonly used carriers, preparation methods, characterization techniques, and recent advancements in the field. The article also discusses the potential of solid dispersion systems in improving the solubility and therapeutic performance of poorly water-soluble drugs
Keywords
Solid dispersion, Solubility, BCS, Polymer
Introduction
×
Any form of dosage has a fundamental characteristic of solubility, meaning that the active compound's properties can be improved by chemical alteration, such as combining weakly soluble compounds with water-soluble carriers. In order to categorize drug substances according to their membrane permeability and aqueous solubility, the Biopharmaceutical Classification System (BCS) was developed in the middle of the 1990s. [1]
Table 1: Drugs are classified as BCS-I to BCS-IV
Classification Property
Solubility
Permeability
BCS-I Highly Soluble
High
High
BCS-II Low Soluble
Low
High
BCS-III Highly Soluble
High
Low
BCS-IV Low Soluble
Low
Low
BCS classifications and criteria:
The BCS classifies drugs into four groups according on their permeability and solubility characteristics: A. BCS class I: high solubility, high permeability: drugs with exceptional absorption properties that are both highly soluble and highly permeable. B. BCS class II: low solubility, high permeability: drugs that can readily pass across biological membranes but have limited solubility in water. C. BCS class III: high solubility, low permeability: drugs with a high solubility in water but struggle to permeable through biological membranes D. BCS class IV: low solubility, low permeability: drugs with limited permeability across biological barriers and poor aqueous solubility. [2]
From the above table we understand that BCS class II drug & BCS Class IV drug have low aqueous solubility. Thus, various solubility enhancement techniques are need to be administered.
Importance of solubility enhancement in drug development
When taken orally, medications that are poorly soluble in water frequently need high dosages to achieve adequate plasma concentrations. Developing new chemical entities and developing substitutes both face the major challenge of low water solubility. At the site of absorption, drugs that is to be absorbed must be present as an aqueous solution. Over 40% of the novel chemical entities (NCEs) created in the pharmaceutical sector are basically water insoluble. These drugs hindered absorption and poor water solubility results in gastrointestinal mucosal toxicity as well as insufficient and inconsistent bioavailability. Solubility is the most crucial rate-limiting factor for oral medications in order to reach the appropriate concentration in the systemic circulation for pharmacological action. For formulation scientists, the solubility issue is a significant obstacle. [3]
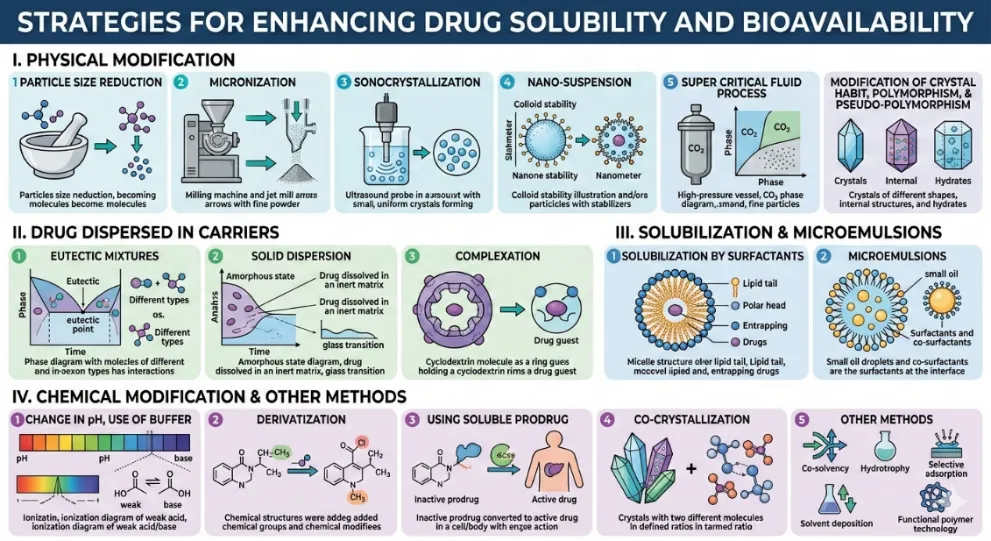
Overview of solubility enhancement techniques [4]
Physical modification
Particle size reduction
Micronization
Sonocrystallization
Nano-suspension
Super critical fluid process
Modification of crystal habit
Polymorphism
Pseudo polymorphism
Drug dispersed in carriers
Eutectic mixtures
Solid dispersion
Complexation
Using complexing agents
Solubilization and microemulsion
Solubilization by surfactants
Micro emulsions
Chemical modification and other modification
Change in pH
Use of buffer
Derivatization
Other methods
Co-crystallization
Co-solvency
Hydrotrophy
Solubilizing agents
Selective adsorption on insoluble carrier
Solvent deposition
Using soluble prodrug
Functional polymer technology
Figure 1: Strategies for enhancing drug solubility and bioavailability obtained from google gemini.
Solid Dispersion:
The term "solid dispersion" describes the dispersion of one or more hydrophobically active substances in a hydrophillic inert carrier in a solid state using various techniques.
The finished product has both a hydrophilic matrix and a hydrophobic drug. [5]
The fundamental idea behind improving a drug's poor solubility with solid dispersion is to completely eliminate the drug's crystalline structure and molecular dispersion in a hydrophilic polymeric carrier. The drug is released as fine colloidal particles when the solid dispersion is exposed to liquids, causing the carrier substance to dissolve. This makes poorly water-soluble drugs more bioavailable by increasing their surface area of dissolution rate. By increasing the porosity and decreasing the size of the particles, the drug within a soluble hydrophilic carrier accelerates the rate of dissolution. Therefore, these drugs bioavailability and adverse effects can be improved by enhancing their drug release profile. [6]
Advantages of solid dispersion [7]
Particles with smaller sizes are produced via solid dispersion, which improves surface area and increases the rate of disintegration. Bioavailability rises as a result.
2. A significant factor in increasing the particles wettability is the carrier employed in the solid dispersion. higher solubility with improved wettability leads to higher bioavailability.
3. Drugs are depicted as supersaturated solutions in solid dispersion, which are thought to be metastable polymorphic forms. Consequently, the drug is presented in an amorphous state, and the particles' solubility is increased.
Disadvantages of Solid Dispersion [7]
Their instability is a major drawback. As they age, they exhibit alterations in crystallinity and a reduction in the rate of dissolution.
Solid dispersions are more susceptible to degradation from temperature and moisture than physical mixes.
Handling challenges due to tackiness.
Limitations of Solid Dispersion [8]
Over the past forty years, solid dispersion has attracted a lot of scholarly interest, but there has been very little commercial use.
Solid dispersion issues include:
the physical and chemical stability of medications and vehicles;
the preparation process;
the repeatability of its physicochemical properties;
the formulation of solid dispersion into dosage forms; and
the scaling up of production operations.
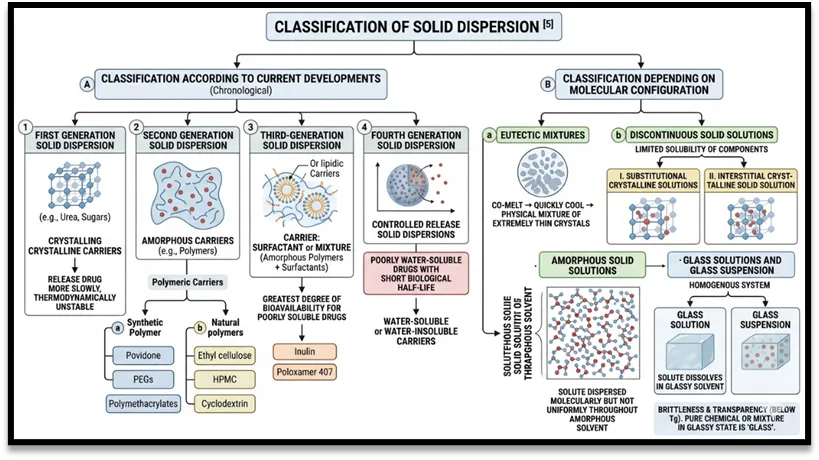
Classification of Solid Dispersion [5]
Figure 2: Classification of Solid Dispersion Picture obtained from Google Gemini
Depending on the molecular configuration, solid dispersions can be divided into the following groups:
Eutectic mixtures: A popular technique for producing solid eutectic mixes is to quickly cool the two components, co-melt to produce a physical mixture of extremely thin crystals.
Discontinuous solid solutions: In these solutions, each component's solubility in the other component is limited. Depending on how the solvates are dispersed throughout the solvent, there are two types of solid solutions:
Substitutional crystalline solutions: Solid solutions that are crystalline are known as substitutional crystalline solutions.
Interstitial crystalline solid solution: In the crystal lattice of nature, the solute molecules take the place of the solvent molecules.
Amorphous solid solutions: The solute molecules in this kind of solution are dispersed molecularly but not uniformly throughout the amorphous solvent.
Glass solutions and glass suspension: A homogenous system known as a glass solution is created when the solute dissolves in the glassy solvent. Transparency and brittleness characterize the glassy state below the glass transition temperature. A pure chemical or a mixture of pure chemicals in their glassy state is referred to as "glass." Solid dispersion classification according to current developments;
First generation solid dispersion: These solid dispersions are made using crystalline carriers. Urea and sugars were the first crystalline carriers used to make solid dispersions. The disadvantage among these is that they release drugs more slowly and are thermodynamically unstable.
Second generation solid dispersion: These solid dispersions are made using amorphous carriers rather than crystalline ones. The drug has been dispersed molecularly within the polymeric carrier. Polymeric carriers fall into two categories:
Synthetic Polymer; such as Povidone, polyethylene glycols, and polymethacrylates are examples of synthetic polymers.
Natural polymers; such as ethyl cellulose, hydroxypropylnethylcellulose, and starch derivatives like cyclodextrin.
Third-generation solid dispersion: In these solid dispersions, the carrier is either a surfactant or a mixture of amorphous polymers and surfactants.
The greatest degree of bioavailability is attained by drugs with poor solubility. Inulin, poloxamer 407, and other surfactants are used in the third-generation solid dispersion [5]
Fourth generation solid dispersion: Researchers refer to these as solid dispersions with controlled release. It comprises drugs with a short biological half-life that is poorly soluble in water. Water-soluble or water-insoluble carriers are utilized. [9]
The Mechanism by which Solubility and Dissolution Rate Enhancement Occurs In Solid Dispersion
By particle size reduction and reduced agglomeration: When a eutectic mixture of a highly soluble carrier and a poorly soluble drugs is exposed to water or gastrointestinal fluid, the soluble carrier dissolves, leaving the drug in a very fine crystalline state that will quickly dissolve. The Noyes-Whitney equation can be used to determine that an increased surface area of the insoluble chemical results in an accelerated dissolving rate and, consequently, increased oral absorption. Similar to this, a solid solution of poorly soluble drugs in a rapidly dissolving carrier dissolves more quickly than a eutectic combination because the drug particles are molecularly dispersed in the carrier in a solid solution, reducing their size to its absolute minimum. [10]
Formation of amorphous structure replacing crystalline structure: Drugs that are poorly soluble in water tend to be more soluble when they are in an amorphous state. Amorphous solids have greater molecular mobility, vapor pressure, dissolving rate, and temporary solubility. Typically, energy is needed to disrupt the crystalline lattice in order to dissolve a crystalline drug. This necessary energy is often thought of as a barrier to the drug's breakdown. The drug is dissolved or molecularly dispersed in a polymeric carrier in solid molecular dispersions, which lack long-range crystalline structure. In this case, the medication is in an amorphous state and has a higher dissolving rate and kinetic solubility (up to many orders of magnitude) than the crystalline drug. [10]
By improving local solubility and wettability of the poorly soluble drug in the solid dispersion matrix: The ability of the polymer carrier to increase the drug's local solubility and wettability also contributes to the improvement in the solubility and dissolution rate of poorly soluble drugs. [10]
Interactions of the drug with Carrier functional groups: Through certain interactions with the drug, the carrier matrix not only enhances the drug's wettability and local solubility but also increases the drug's aqueous solubility and rate of dissolution. [10]
The intermolecular hydrogen bonding: According to Konno and Taylor, felodipine solid dispersions with polyvinylpyrrolidone (PVP), hydroxypropyl methylcellulose acetate succinate (HPMCAS), and hydroxypropyl methylcellulose (HPMC) exhibited hydrogen bonding interactions, allowing all polymers to preserve the drug's amorphous state even at low excipient concentrations. [10]
By elevating the Tg of the solid dispersion mixtures: Polymeric carriers have been demonstrated to delay amorphous drug crystallization by raising the viscosity and glass transition temperatures (Tg) of the polymer-drug mixtures in addition to developing molecular interactions with the drug molecule.
The temperature below which the amorphous mixture is in a glassy state and coordinated molecular motion becomes extremely sluggish and constrained is known as the glass transition temperature. [10]
Inhibited drug precipitation from supersaturated solution: It is feasible to create a solution with a with a drug concentration significantly higher than the solubility of the crystalline drug due to the increased kinetic solubility of the drug's amorphous form. The drug tends to precipitate out with time, reaching a substantially lower equilibrium solubility. The presence of dissolved carrier may also inhibit the precipitation of the drug from the supersaturated solution. [10]
By formation of Metastable drug polymorphous with higher solubility and dissolution rate: By creating metastable crystalline polymorphs of the drug substance that have greater solubility than the crystalline drug alone, polymeric carriers may still be able to increase the drug's solubility and rate of dissolution even in situations where they are unable to produce the full amorphous state of the drug.[10]
Compound or Complex formation: In an aqueous solution, the drug and matrix interact intensely to produce complexes, such as cyclodextrins. Dissolution enhancement requires a low association constant. When a low or intermediate fraction of carrier is used to prepare a solid dispersion, the creation of a soluble complex may occur. The creation of a solid solution may facilitate drug dissolution when a large fraction of carrier is used. [11]
5. Higher Porosity Particles: It has been discovered that particles in solid dispersions have a higher level of porosity. The carrier characteristics also affect the rise in porosity; for example, solid dispersions including linear polymers generate larger and more porous particles than those containing reticular polymers, which leads to a higher rate of dissolution. Additionally, the drug release profile is accelerated by the solid dispersion particles' enhanced porosity. [12]
Selection of the carrier:
Since the dissolving rate of one component from the surface is influenced by the other component in a multiple component mixture, the choice of carrier has an impact on the dispersed drug's dissolution characteristics. The drug is therefore released from the matrix more quickly when a water-soluble carrier is used. The drug is released from the matrix more slowly when the carrier is insoluble or weakly soluble. A drug can be released from the matrix more quickly if the active ingredient is a small part of the dispersion. [13]
Properties of a carrier that are appropriate for solid dispersions.
High water solubility increases dissolution and wettability.
Polyglycolized glycerides: 44/14, 50/13, and 62/05 Gelucire
Acids: Phosphoric acid, succinic acid, and citric acid
Miscellaneous: Microcrystalline cellulose, silica gel, sodium chloride, di calcium phosphate, and skim milk.
Method of Preparation:
Different methods that are used for the preparation of the solid dispersion are enlist below:
Fusion / Melting method
Solvent method
Melting solvent method (melt evaporation)
Melt extrusion methods
Lyophilization techniques
Melt agglomeration Process
The use of surfactant
Electrospinning
Super Critical Fluid (Scf) technology
Dropping method solution:
Figure 3: Methods of preparation of solid dispersion picture obtained from google gemini.
Fusion / Melting method: A physical mixture of a drug and a hydrophilic carrier is prepared using the melting or fusing process, which involves heating the mixture directly until it melts. After that, the melted fluid is quickly hardened while being vigorously stirred in an ice bath. The final solid mass is ground up, sieved, and crushed.
The process of pouring the homogenous melt in the shape of a thin layer onto a stainless-steel plate and cooling it with flowing water or air on the other side of the plate has undergone numerous changes. Additionally, it is frequently possible to achieve a super-saturation of a medication in a system by quickly cooling the melt from a high temperature.
This approach's primary benefits are its affordability and ease of use. The procedure is only used when,
The drug and matrix are compatible and combine properly at the heating temperature, which is one of its drawbacks. When the medication and matrix are incompatible, the heated mixture may show two liquid phases or suspension, which leads to an inhomogeneous solid dispersion. Surfactants can be used to avoid this issue.
When the drug-matrix miscibility shifts during cooling, another issue could occur. Phase separation may happen in this situation. In fact, it was found that rapid cooling produced amorphous solid dispersions, while delayed cooling produced crystalline medication.
At high temperatures, many substance - drugs or carriers may break down during the fusion process. [13]
Solvent method: Preparing a solution with the drug and matrix material is the first step in the solvent technique. In order to create a solid dispersion, the solvent or solvents must be eliminated in the second phase. The best dissolving properties result from mixing at the molecular level.
The primary benefit of the solvent approach is that, due to the comparatively low temperatures needed for the evaporation of organic solvents, thermal breakdown of drugs or carriers can be avoided. The first problem is combining the medication and matrix in a single solution, which is challenging when their polarity differs greatly. The drug and matrix must be disseminated as finely as feasible in the solvent to reduce the drug particle size in the solid dispersion; ideally, the drug and matrix material are dissolved in a single solution. The drug and matrix material are dissolved in water using low drug concentrations, however this procedure is costly and unfeasible because it necessitates the evaporation of enormous volumes of solvent. The drug's water solubility is significantly increased by solubilizers like cyclodextrins or surfactants like Tween80®.reventing phase separation, such as drug or matrix crystallization, during solvent removal is the second hurdle in the solvent approach. In order to eliminate any remaining solvent, the resulting solid dispersion is then frequently kept in vacuum desiccators. Spray drying is an additional drying method. These factors make hot melt extrusion the preferred technique for creating solid dispersions nowadays.[13]
Melting solvent method (melt evaporation): In order to prepare solid dispersions, the drug must be dissolved in an appropriate liquid solvent. The solution is then added straight to the melted polyethylene glycol which is subsequently evaporated until a clear, solvent-free film remains. The film is subsequently dried until its weight remains consistent. The drug's polymorphic form, which precipitates as a solid dispersion, may be impacted by the liquid solvent utilized. This process combines the special benefits of solvent evaporation and fusing. Practically speaking, it is only applicable to medications with low therapeutic doses, such as less than 50 mg, and is especially helpful for drugs with high melting points or thermolabile properties.[13]
Melt extrusion method: This approach uses a co-rotating twin-screw extruder to prepare solid dispersion, which is made up of the active component and carrier. Melted and homogenized at the same time, the drug/carrier mixture is extruded and formed into tablets, granules, pellets, sheets, sticks, or powder. Melted and homogenized at the same time, the drug/carrier mixture is extruded and formed into tablets, granules, pellets, sheets, sticks, or powder. In addition, HME has a number of benefits over conventional pharmaceutical processing methods, such as the lack of solvents, minimal processing stages, continuous operation, low temperature, and short residence time that shields the drug-carrier mixture from thermal.
Some drawbacks include:
strong shear pressures may cause the extruder's local temperature to rise, which could cause issues for heat-sensitive materials; and
miscibility of the drug and carrier matrix could be an issue, similar to the old fusion approach. [13]
Lyophilization Technique (Freeze-drying): In order to create a lyophilized molecular dispersion, the drug and carrier are co-dissolved in a shared solvent, frozen, and sublimated. This process is known as lyophilization. This method was suggested as a substitute for solvent evaporation. Freeze drying has the advantage of minimizing the possibility of phase separation as soon as the solution is vitrified and subjecting the drug to less thermal stress during the development of the solid dispersion. Spray-freeze drying is an even more promising drying method. The frozen droplets are then lyophilized after the solvent is poured into liquid nitrogen or cold, dry air. Due to vast surface area and direct contact with the cooling agent, condensation occurs even more quickly, reducing the possibility of phase separation. Additionally, spray freeze drying provides the opportunity to modify the particle's size to make it appropriate for additional processing or uses like nasal or pulmonary delivery. [13]
Melt Agglomeration Process: This method has been used to create solid dispersions in which the binder serves as a carrier. Additionally, solid dispersions are made by either employing a high shear mixer to spray a drug dispersion in molten binder on the heated excipient (spray-on process) or heating the binder, drug, and excipient to a temperature higher than the binder's melting point (melt-in procedure). Because it is simpler to regulate the temperature and because the agglomerates can have a larger binder concentration, the rotary processor may be better than the high melt agglomeration. Due to the immersion process of agglomeration formation and growth, it has been discovered that the melt-in method yields higher dissolution rates than the spray-on method using PEG 3000, poloxamer 188, and gelucire 50/13. Additionally, the drug is distributed uniformly throughout the agglomerate as a result of the melt-in process. While small particles promote total adherence to the material of bowl, soon after melting due to their dispersion and coalescence, larger particles cause agglomerates to densify. [13]
The use of surfactant: The surfactant systems effectiveness in solubilization is crucial. Surfactants' hydrophobicity, surface charge, and other critical characteristics that control interfacial processes including flocculation/dispersion, floating, wetting, solubilization, detergency, improved oil recovery, and corrosion inhibition can all be altered by adsorption on solid surfaces. Additionally, it has been reported that surfactants produce plasticization and solvation, that indicates a decrease in the glass transition temperature, the melting point of active pharmaceutical substances, and the combined glass transition temperature of solid dispersions. Because of these special qualities, researchers are interested in using surfactants to make solid dispersions. [13]
Electrospinning: The method of electrospinning involves delivering a polymeric fluid stream solution or molten via a millimeter-scale nozzle to create solid fibers. In this procedure, a conducting capillary attached to a reservoir holding a polymer solution or melt and a conductive collection screen is subjected to a strong electrostatic field. Charge species build up on the surface of a pendant drop when the electrostatic field intensity is increased up to but not above a critical value; this destabilizes the hemispherical shape and transforms it into a conical shape, also referred to as Taylor's cone. In order to release the charge accumulated on the pendant drop's surface, a charged polymer jet is expelled from the cone's apex beyond the critical value. [13]
Super Critical Fluid (Scf) Technology: Carbon dioxide is employed as an antisolvent for the solute but as a solvent for the organic solvent in supercritical fluid antisolvent techniques. Aerosol solvent extraction system, precipitation with a compressed fluid or gas anti-solvent, solution improved dispersion by supercritical
antisolvent and fluids. The drug particles may recrystallize at significantly smaller particle sizes after being dissolved in SCF. Drug particles can be micronized within specific particle size ranges, frequently to sub-micron levels, thanks to the flexibility and accuracy provided by SCF techniques.
Even though a tiny quantity of carbon dioxide is still retained inside the polymer, using supercritical carbon dioxide is beneficial since it is much easier to remove from the polymeric materials after the process is finished; the patient is not at risk. Additionally, it is possible to take use of carbon dioxide's capacity to plasticize and expand polymers at temperatures close to room temperature. Additionally, by lowering the melting temperature of the dispersed active ingredient, supercritical fluids are employed to lower the temperature of the melt dispersion process.
SCFs are appealing for pharmacological research because of their low operating conditions (temperature and pressure). [13]
Dropping method solution: The dropping method is a novel technique for creating spherical particles from various substances to aid in their crystallization
from those solid dispersions that have been melted. Some of the challenges inherent in the methodologies may be overcome using this strategy. A solid dispersion of a melting drug-carrier mixture is pipetted and deposited onto a plate for laboratory-scale production, where it hardens into spherical particles. Using carriers that solidify at ambient temperature could facilitate the process of dropping. The dropping method provides a higher dissolving rate in addition to simplifying the manufacturing process. Since it doesn't use organic solvents, it doesn't have any issues with solvent evaporation. [6]
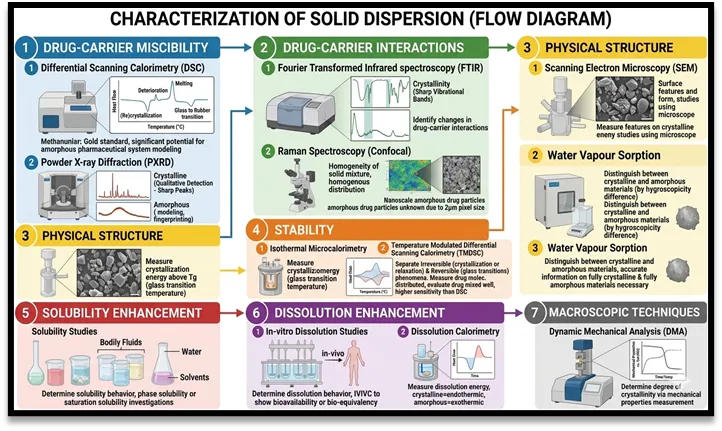
Characterization of solid dispersion: [12]
Figure 4: Characterization of solid dispersion pictorial flow diagram obtained from google gemini.
Drug-carrier Miscibility
Differential Scanning Calorimetry (DSC): This method is frequently employed to determine the quantity of crystalline material. This method measures the amount of energy required to heat samples at a steady pace. The temperatures at which thermal events take place can be determined by DSC. Thermal events include melting, deterioration, (re)crystallization, and the change from glass to rubber. Additionally, it is possible to quantify the melting and (re)crystallization energy. The amount of crystalline material can be determined using the melting energy.
Powder X-ray Diffraction (PXRD): X-ray diffraction serves as gold standard for solid-state pharmaceutical material characterization. It has demonstrated significant potential for amorphous pharmaceutical system modeling, measurement, and fingerprinting. Single-crystal and powder X-ray diffraction are the two main types of X-ray diffraction. [16]
Material having long range order can be qualitatively detected using powder X-ray diffraction. More crystalline material is indicated by sharper diffraction peaks.
Drug-carrier Interactions
Fourier Transformed Infrared spectroscopy (FTIR): This method is used to identify changes in the energy distribution of drug-carrier interactions. Crystallinity is indicated by sharp vibrational bands.
Raman Spectroscopy (Confocal): The homogeneity of the solid mixture is measured using Confocal Raman spectroscopy. It is explained
that a homogenous distribution was indicated by a drug content standard deviation of less than 10%. Due to the 2μm pixel size. The existence of nanoscale amorphous drug particles is still unknown.
Physical Structure
Scanning Electron Microscopy (SEM): Scanning electron microscopy was used to examine the surface features and form of solid dispersion systems.
studies using microscope.
Water Vapour Sorption: When the hygroscopicity is varied, water vapor sorption can be utilized to distinguish between crystalline and amorphous materials.
Stability
Isothermal Microcalorimetry: When an amorphous material is heated above its glass transition temperature (Tg), the crystallization energy is measured.
Temperature Modulated Differential Scanning Calorimetry (TMDSC): This method can be used to measure the amount of drug that is molecularly distributed and determine the proportion of drug that diffuses as separate molecules since its sensitivity is higher than that of DSC. This can also be used to evaluate how well a drug is mixed. In amorphous materials, modulation allows for the separation of irreversible (crystallization or relaxation) and reversible (glass transitions) phenomena.
Water Vapour Sorption: Water vapor sorption can be utilized to distinguish between crystalline and amorphous materials if hygroscopicity is different. Accurate information on the hygroscopicity of both fully crystalline and fully amorphous materials is necessary.
Solubility Enhancement
Solubility Studies: Solubility studies are conducted to determine the solid dispersion system's solubility behavior in various solvent types,
bodily fluids and systems. Phase solubility or saturation solubility investigations can accomplish this.
Dissolution Enhancement
In-vitro Dissolution Studies: To determine dissolution behavior, in-vitro dissolution tests are conducted. Through in-vitro/in-vivo correlation (IVIVC), the in-vitro dissolution research can be utilized to show the drug product's bioavailability or bio-equivalency.
Dissolution Calorimetry: The energy of dissolution, which depends on the sample's crystallinity, is measured by dissolution calorimetry. Crystalline materials often dissolve endothermically, while amorphous materials dissolve exothermically.
Macroscopic Techniques
Dynamic Mechanical Analysis (DMA): The degree of crystallinity can be determined via (DMA), which measures mechanical properties.
CONCLUSION
It is evident from this review that one of the most sophisticated methods for addressing the issue of poorly water-soluble medication solubility is solid dispersion technology. Therefore, it is essential to look into the physio-chemical characteristics of the drug and carrier that can work best together before creating a novel solid dispersion system for a particular drug. Additionally, the ratio of carrier to drug and the preparation technique are important factors in improving the drug's solubility and rate of dissolution. In order to accomplish this purpose, we have tried to put everything in order in this article.
REFERENCES
Jindal K. Review on solubility: A mandatory tool for pharmaceuticals. International Research Journal of Pharmacy. 2017;8(11):11-5.
Aru PB, Kale SR, Hiwase RD, Manekar SR, Deshmukh SP, Deshmukh NB. Review on biopharmaceutics classification system (BCS). European Journal of Biomedical and Pharmaceutical Sciences. 2023;10(9):XX–XX.
Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. International Scholarly Research Notices. 2012;2012(1):195727.
Jindal K. Review on solubility: A mandatory tool for pharmaceuticals. International Research Journal of Pharmacy. 2017;8(11):11-5.
Kaur S, Bhatti F, Amisha, Kumar A. A review on solid dispersion. J Res Appl Sci Biotechnol. 2024;3(2):295–300.
Sarkate G, Sarode A, Selmokar O, Harpale P, Oswal R. A review on solid dispersion: A technology for improving bioavailability. Int J Pharm Res Appl. 2022 Jul-Aug;7(4):1020-1030.
Kumar B. Solid dispersion-a review. PharmaTutor. 2017 Feb 1;5(2):24-9.
Das SK, Roy S, Kalimuthu Y, Khanam J, Nanda A. Solid dispersions: an approach to enhance the bioavailability of poorly water-soluble drugs. International journal of pharmacology and pharmaceutical technology. 2012;1(1):37-46.
Totre A, Chaurasia G. A Review on Solid Dispersion Techniques: A Exclusive Approach for Enhancing Drug Solubility and Bioavailability. Int J Multidiscip Res. 2025 May-Jun;7(3).
Sapkal S, Babhulkar M, Rathi A, Mehetre G, Narkhede M. An overview on the mechanisms of solubility and dissolution rate enhancement in solid dispersion. Int J Pharm Tech Res. 2013;5(1):31-9.
Ramesh V, Meenakshi S, Jyothirmayee N, Bullebbai M, Noorjahan SK, Rajeswari G, Nagesh Babu G, Madhavi D. Enhancement of solubility, dissolution rate and bioavailability of BCS Class II Drugs. Int J Pharma Chem Res. 2016 Apr;2(2):80-95.
Mir KB, Khan NA. Solid dispersion: Overview of the technology. International journal of pharmaceutical sciences and research. 2017 Jun 1;8(6):2378-87.
Sridhar I, Doshi A, Joshi B, Wankhede V, Doshi J. Solid dispersions: an approach to enhance solubility of poorly water soluble drug. J Sci Innov Res. 2013;2(3):685-94.
Sharma R, Mazumder R, Sharma A, Verma P. A review on: Solid dispersion. International Journal of Pharmacy & Life Sciences. 2013 Jul 1;4(7).
Ramesh V, Meenakshi S, Jyothirmayee N, Bullebbai M, Noorjahan SK, Rajeswari G, Nagesh Babu G, Madhavi D. Enhancement of solubility, dissolution rate and bioavailability of BCS Class II Drugs. Int J Pharma Chem Res. 2016 Apr;2(2):80-95.
Liu X, Feng X, Williams III RO, Zhang F. Characterization of amorphous solid dispersions. Journal of Pharmaceutical Investigation. 2018 Jan;48(1):19-41.
Reference
Jindal K. Review on solubility: A mandatory tool for pharmaceuticals. International Research Journal of Pharmacy. 2017;8(11):11-5.
Aru PB, Kale SR, Hiwase RD, Manekar SR, Deshmukh SP, Deshmukh NB. Review on biopharmaceutics classification system (BCS). European Journal of Biomedical and Pharmaceutical Sciences. 2023;10(9):XX–XX.
Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. International Scholarly Research Notices. 2012;2012(1):195727.
Jindal K. Review on solubility: A mandatory tool for pharmaceuticals. International Research Journal of Pharmacy. 2017;8(11):11-5.
Kaur S, Bhatti F, Amisha, Kumar A. A review on solid dispersion. J Res Appl Sci Biotechnol. 2024;3(2):295–300.
Sarkate G, Sarode A, Selmokar O, Harpale P, Oswal R. A review on solid dispersion: A technology for improving bioavailability. Int J Pharm Res Appl. 2022 Jul-Aug;7(4):1020-1030.
Kumar B. Solid dispersion-a review. PharmaTutor. 2017 Feb 1;5(2):24-9.
Das SK, Roy S, Kalimuthu Y, Khanam J, Nanda A. Solid dispersions: an approach to enhance the bioavailability of poorly water-soluble drugs. International journal of pharmacology and pharmaceutical technology. 2012;1(1):37-46.
Totre A, Chaurasia G. A Review on Solid Dispersion Techniques: A Exclusive Approach for Enhancing Drug Solubility and Bioavailability. Int J Multidiscip Res. 2025 May-Jun;7(3).
Sapkal S, Babhulkar M, Rathi A, Mehetre G, Narkhede M. An overview on the mechanisms of solubility and dissolution rate enhancement in solid dispersion. Int J Pharm Tech Res. 2013;5(1):31-9.
Ramesh V, Meenakshi S, Jyothirmayee N, Bullebbai M, Noorjahan SK, Rajeswari G, Nagesh Babu G, Madhavi D. Enhancement of solubility, dissolution rate and bioavailability of BCS Class II Drugs. Int J Pharma Chem Res. 2016 Apr;2(2):80-95.
Mir KB, Khan NA. Solid dispersion: Overview of the technology. International journal of pharmaceutical sciences and research. 2017 Jun 1;8(6):2378-87.
Sridhar I, Doshi A, Joshi B, Wankhede V, Doshi J. Solid dispersions: an approach to enhance solubility of poorly water soluble drug. J Sci Innov Res. 2013;2(3):685-94.
Sharma R, Mazumder R, Sharma A, Verma P. A review on: Solid dispersion. International Journal of Pharmacy & Life Sciences. 2013 Jul 1;4(7).
Ramesh V, Meenakshi S, Jyothirmayee N, Bullebbai M, Noorjahan SK, Rajeswari G, Nagesh Babu G, Madhavi D. Enhancement of solubility, dissolution rate and bioavailability of BCS Class II Drugs. Int J Pharma Chem Res. 2016 Apr;2(2):80-95.
Liu X, Feng X, Williams III RO, Zhang F. Characterization of amorphous solid dispersions. Journal of Pharmaceutical Investigation. 2018 Jan;48(1):19-41.
Rutuja Pote
Corresponding author
Shivnagar Vidya Prasarak Mandal's College of Pharmacy, Malegoan BK Baramati.
Dr. T. Deshmukh
Co-author
Shivnagar Vidya Prasarak Mandal's College of Pharmacy, Malegoan BK Baramati.
Rutuja Pote, Dr. T. Deshmukh, A Comprehensive Review On Solid Dispersion Techniques For Solubility Enhancement Of Drugs, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5873-5886, https://doi.org/10.5281/zenodo.20810655




 10.5281/zenodo.20810655
10.5281/zenodo.20810655