
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Quality Assurance, S.G.S.P.S Institute of Pharmacy Kaulkhed, Akola
A simple, rapid, precise, and accurate reverse-phase high-performance liquid chromatographic (RP-HPLC) method was developed and validated for the simultaneous determination of Atorvastatin Calcium and Ezetimibe in bulk drug and its pharmaceutical formulation. The chromatographic separation was achieved on a C18 column using a mobile phase composed of phosphate buffer (pH 3.5) and acetonitrile in the ratio of 40:60 v/v, at a flow rate of 1.0 mL/min with isocratic elution. The detection was carried out at 246 nm using a UV detector. The retention times for Atorvastatin and Ezetimibe were found to be 3.2 min and 5.1 min, respectively. The method was validated as per ICH Q2(R1) guidelines for linearity, accuracy, precision, specificity, robustness, LOD, and LOQ. Linearity was observed in the concentration range of 10–50 ?g/mL for Atorvastatin (r² = 0.9996) and 5–25 ?g/mL for Ezetimibe (r² = 0.9998). The %RSD for intraday and interday precision was <2%. The mean percentage recoveries were found to be 99.8% for Atorvastatin and 99.5% for Ezetimibe. The LOD and LOQ were 0.32 ?g/mL and 0.98 ?g/mL for Atorvastatin, and 0.18 ?g/mL and 0.55 ?g/mL for Ezetimibe, respectively. The developed method was successfully applied for the routine quantitative analysis of Atorvastatin and Ezetimibe in combined tablet dosage form without interference from excipients. The results indicate that the proposed RP-HPLC method is suitable, reliable, and economical for simultaneous estimation of both drugs in bulk and pharmaceutical formulations.
Analytical methods development and validation play important roles in the discovery, development, and manufacture of pharmaceuticals. Pharmaceutical products formulated with more than one drug, typically referred to as combination products, are intended to meet previously unmet patients need by combining the therapeutic effects of two or more drugs in one product. These combination products can present daunting challenges to the analytical chemist responsible for the development and validation of analytical methods. This presentation will discuss the development and validation of analytical method (Spectrophotometric, High performance liquid chromatography (HPLC), & High-performance thin layer chromatography (HPTLC)) for drug products containing more than one active ingredient. The official test methods that result from these processes are used by quality control laboratories to ensure the identity, purity, potency, and performance of drug products. The number of drugs introduced into the market is increasing every year. These drugs may be either new entities or partial structural modification of the existing one. Very often there is a time lag from the date of introduction of a drug into the market to the date of its inclusion in pharmacopoeias. This happens because of the possible uncertainties in the continuous and wider usage of these drugs, reports of new toxicities (resulting in their withdrawal from the market), development of patient resistance and introduction of better drugs by competitors. Under these conditions, standards and analytical procedures for these drugs may not be available in the pharmacopoeias. It becomes necessary, therefore to develop newer analytical methods for such drugs.
BASIC CRITERIA FOR NEW METHOD DEVELOPMENT OF DRUG
ANALYSIS:
1. The drug or drug combination may not be official in any pharmacopoeias.
2. A proper analytical procedure for the drug may not be available in the literature due to patent regulations.
3. Analytical methods may not be available for the drug in the form of a formulation due to the interference caused by the formulation excipients.
4. Analytical methods for the quantitation of the drug in biological fluids may not be available.
5. Analytical methods for a drug in combination with other drugs may not be available.
6. The existing analytical procedures may require expensive reagents and solvents. It may also involve cumbersome extraction and separation procedures and these may not be reliable.
CHOICE OF ANALYTICAL METHOD
The analytical method chosen should be having all the ideal characteristics and the most important being that it should be less time consuming. This is about the method development relatively simple task but in case of combined dosage form the scenario is different as the properties of the one drug may hamper the properties of others. These properties may include solubility, shifting of λmax, overlapping of the absorbance, etc. But the various sophisticated analytical instruments are now overcoming these problems.A vital first step in any quantitative analysis is the selection of method. This will require careful consideration of following criteria.
1. The type of analysis required.
2. Problems arising from the nature of material to be investigated.
3. Possible interference from components of the material other than those of interest.
4. The concentration range, which needs to be investigated.
5. The accuracy required.
6. The facilities available.
7. The time required to complete the analysis.
8. The number of analysis of similar types, which have to be performed.
Finally, the choice of method is always governed by complexity of sample as well as number of component.
SPECTROPHOTOMETRIC METHODS
Absorption spectroscopy is one of the most useful and widely used tools available to the analyte for quantitative analysis. The relation between the concentration of analyte and the amount of light absorbed is the basis of most analytical applications of molecular spectroscopy. This method of analysis is gaining importance due to simple, rapid, precise, highly accurate and less time consuming. Spectrophotometric multi-component analysis can be applied where the spectra of drugs overlaps. It utilizes measurement of the intensity of electromagnetic Radiation emitted or absorbed by the analyte.
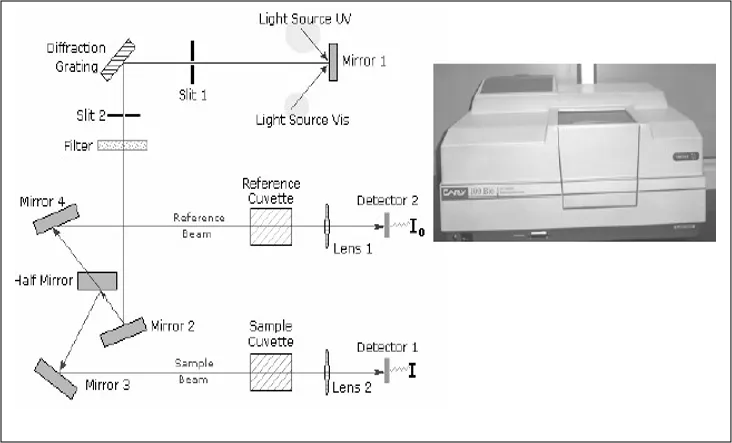
UV-visible spectrophotometer and its flow diagram
REVIEW OF LITERATURE
Review should be based on assimilation of data from various sources for the purpose of establishing a new concept. A preliminary survey of literature for suitable method development for newer drugs has been made.Review of literature reveals that extensive of work has been carried out for the routine analysis of drugs and pharmaceuticals in recently marketed as well as existing formulations and bulk drugs. Development of high performance liquid chromatographic method can be successfully employed for the analysis of formulations. Some literature survey of the present study is under.
1.Sandeep Bolla et. al, (2025) Studied Stability indicating liquid chromatographic method for the simultaneous estimation of Colchicine and Rosuvastatin in combination. This study focuses on developing and validating a robust, reliable, and stability indicating reverse-phase high-performance liquid chromatography (RP-HPLC) method for the simultaneous quantification of Colchicine and Rosuvastatin in bulk and synthetic mixture. The developed method is designed to meet International Council for Harmonization (ICH) guidelines for stability-indicating assays. The mobile phase consisted of buffer (pH adjusted to 4.0 with ortho-phosphoric acid): methanol:acetonitrile: in a 50:30:20 (v/v/v) ratio, and separation was performed on a Shim-pack ODS C18 (250 mm × 4.6 mm, 5 µm) column. The method was validated for specificity, linearity, precision, accuracy, and robustness. Forced degradation studies were performed under acidic, basic, oxidative, photolytic, and thermal stress conditions. [26]
2. Sakshi Hadawale et. al, (2024) Studied Simultaneous determination of Rosuvastatin calcium and Bempedoic acid in fixed-dose combination pharmaceutical dosage forms using RP-HPLC method. A precise, simple, and, accurate method for simultaneous estimation of Rosuvastatin calcium and Bempedoic acid was developed by Reversed Phase High-Performance Liquid Chromatographic (RP-HPLC). The validation of the developed method is done as per ICH guidelines. The method was developed on Waters HPLC using a Kromasil 100-5-C18 (4.6 × 250 mm) column with a detection wavelength of 220 nm. The mobile phase consists of 0.1 % OPA in water: Acetonitrile(50:50 v/v). The retention time for Rosuvastatin calcium and Bempedoic acid was found as 5.4 and 10.7 min respectively. [27]
3. Verma N, Singh et. al, (2023): Verma and Singh conducted a study on the RP-HPLC method development and validation for the simultaneous determination of antimuscarinic drugs in herbal formulations. The researchers aimed to establish are liable and accurate method for quantifying these drugs in complex herbal matrices used in traditional medicine. The study involved optimizing the chromatographic conditions and mobile phase composition to achieve good separation and resolution of the target analytes in the herbal formulations. The developed method was validated for its accuracy, precision, and sensitivity. [28]
4.Gore et. al, (2022) reviewed that the Gliptin, commonly known as dipeptidylpeptidase-4, is a type of anti-diabetic medication. DPP-4 (dipeptidyl peptidase4) inhibitors are a type of anti-hyperglycemic drug that works by inhibiting the enzyme DPP-4 and increasing the biological activity of the "incretin" hormones (glucagon-like peptide-1 [GLP-1] and glucose-dependent insulinotropic polypeptide). The gliptin class inhibitors Sitagliptin, Saxagliptin, Alogliptin, Linagliptin, and Vildagliptin are used to treat type 2 diabetes mellitus by reducing the breakdown of incretin hormones like GLP1. The reviewer demonstrated the development and validation of HPLC methods for Saxagliptin, Linagliptin, and Vildagliptin, Sitagliptin, and Alogliptin alone and in combination in this work. The pharmacokinetic and pharmacodynamic properties of gliptins were also highlighted by the reviewer. [29]
5.Sammi Akter et. al, (2022) : An accurate, precise and cost effective HPLC method was developed that is not appeared in pharmacopoeia for estimation of Sitagliptin. Separation of the drug was achieved on a C18 column using a mobile phase consisting of phosphate buffer and acetonitrile in the ratio of 60: 40 (v/v). The flow rate was1mL/min and detection wavelength was 254 nm. The linearity was observed in the concentration of 0.05, 0.20, 0.22, 0.25, 0.27, 0.29 mg/mL with a correlation coefficient (R2) of 0.999. The retention time of Sitagliptin was 5.2±0.03 min. The proposed method was validated as per the ICH guidelines for the parameters: Linearity, Accuracy, Precision, Robustness, and Specificity etc. This method can be employed for routine quality control analysis of Sitagliptin in tablet dosage form. [30]
6. Andreas Vrachas, et. al, (2022) Studied that Cetrimide (CE) is a quaternary ammonium compound and a cationic surfactant, which can be used as an antiseptic and preservative in various formulations. Antiseptic solutions of Cetrimide are available in combination with Chlorhexidine Gluconate (CHG) for external use. Chlorhexidine is abiguanide with high microbicidal activity and is widely known as a skin disinfectant. The present work displays the development and validation of an RP-HPLC isocratic method for the simultaneous determination of CE and CHG. The method consists of a Hypersil SAS C1 (4.6 × 250 mm) 5 µm column, with a mobile phase of 85%/15% v/v MeOH-NaH2PO4 ·H2O, aqueous solution. In addition, 0.2% of triethylamine (Et3N) was added to the buffer for the confrontation of peak tailing, and then the pH was adjusted to 3.0 with ortho-phosphoric acid (H3PO4). The flow rate was set at 1 mL/min, and adequate detection was achieved with a diode array detector (PDA) at 205 nm. The method was successfully validated according to ICH guidelines for specificity, linearity, accuracy, precision and stability for sample and standard solutions. In addition, the robustness of the method was evaluated through statistical and graphical analysis, using a fractional factorial experimental design. [31]
AIM & OBJECTIVES
AIM: A Research On RP-HPLC Method Development & Validation for The Simultaneous Estimation of Atorvastatin & Ezetimibe In Bulk & It’s Pharmaceutical Dosage Form
OBJEVTIVES:
To select suitable drug ie Atorvastatin & Ezetimibe and drug formulation
PLAN OF WORK
Procurement of pure drug samples of Atorvastatin & Ezetimibe and their marketed formulation in combination. Analysis of Pure sample by reported methods. Trial of the instrumental methods on pure drug samples which includes the following steps
SPECTROPHOTOMETRY
Determination of scanning wavelength
HIGH PERFORMANCE LIQUID CHROMATOGRAPHY
Confirm reproducibility and accuracy. The efforts will also be made to develop the most reliable and highly sensitive High Performance Liquid Chromatographic method for the combination, The steps in Method development will as follows-
PARAMETER FOR VALIDATION TO BE STUDIES
DRUG PROFILE
ATORVASTATIN
Structure:
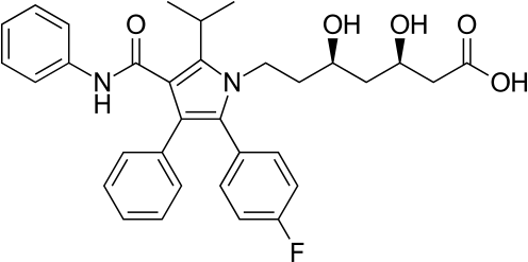
Chemical Name:- (3R,5R)-7-[2-(4-Fluorophenyl)-3-phenyl-4-(phenylcarbamoyl)-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoic acid
Description:- A white to off- white powder
Molecular formula:- C33H35FN2O5
Molecular weight:- 558.650 g/mol
Melting point:- 159-161 0C
Solubility :- It is very slightly soluble in water and slightly soluble in ethanol but is freely soluble in methanol.
Category:- Atorvastatin is an HMG-CoA reductase inhibitor used to lower lipid level and reduce the risk of cardiovascular disease including myocardial infarction and stroke. Mechanism of action:- Atorvastatin is a statin medication and a competitive inhibitor of the enzyme HMG-CoA (3-hydroxy-3-methylglutaryl coenzyme A) reductase, which catalyzes the conversion of HMG-CoA to mevalonate, an early rate-limiting step in cholesterol biosynthesis. Atorvastatin acts primarily in the liver, where decreased hepatic cholesterol concentrations stimulate the upregulation of hepatic low-density lipoprotein (LDL) receptors, which increases hepatic uptake of LDL. Atorvastatin also reduces Very-Low-Density Lipoprotein-Cholesterol (VLDL-C), serum triglycerides (TG) and Intermediate Density Lipoproteins (IDL), as well as the number of apolipoprotein B (apo B) containing particles, but increases High-Density Lipoprotein Cholesterol (HDL-C). In vitro and in vivo animal studies also demonstrate that atorvastatin exerts vasculoprotective effects independent of its lipid-lowering properties, also known as the pleiotropic effects of statins. These effects include improvement in endothelial function, enhanced stability of atherosclerotic plaques, reduced oxidative stress and inflammation, and inhibition of the thrombogenic response. Statins were also found to bind allosterically to β2 integrin function associated antigen-1 (LFA-1), which plays an essential role in leukocyte trafficking and T cell activation.
MATERIALS & METHODS
Materials
The reliability and scientific validity of an analytical method depend largely on the appropriate selection of materials and the systematic application of well-defined experimental procedures. In HPLC method development, careful consideration of the quality of reagents, instrumentation, and analytical conditions is essential to ensure reproducible, accurate, and precise results. The materials employed must meet suitable purity standards, while the experimental methods should be designed in accordance with established regulatory guidelines to minimize analytical variability and bias.The drug used for present investigation was obtained from MG lab Hyderabad as gift sample.
Table No.01:Details of API
|
Sr. No. |
Drug |
Supplied by |
Quantity |
Purity (Assay) |
|
01 |
Atorvastatin |
MGLab India |
10g |
99.3 |
|
02 |
Ezetimibe |
MGLab India |
10g |
99.7 |
Marketed Preparation:
Table No.02:Details of Marketed preparation
|
Sr. No. |
Brand name |
Mfg.by |
Content |
Quantity |
|
01 |
Storvas EZ |
Sun Pharmaceutical Industries Ltd |
Atorvastatin& Ezetimibe |
10mg&10 mg |
METHODS & PROCEDURE
Previous to commenced the experimental work it is necessary to determine the different physical and chemical property of the drug which provide information regarding the purity and nature of drug. This will help in selection of solvents and procedure for the robust and stable analytical method development. Atorvastatin & Ezetimibe
Atorvastatin (ATOR) & Ezetimibe (EZTB) were selected as model drug candidate for method development and validation. The drugs were kindly gifted from Pharmaceutical industry India. The procured drug was analyzed for different physical properties viz. color, odor, melting point, etc.
The physico-chemical characterization of drug molecule is important with regard to its
purity, identification in development and validation of analytical method. The various tools used for characterization of drug molecules include melting point, UV spectroscopy, solubility study, etc.
As a first step of method development solubility of both drugs was tasted in different solvents to obtain a common solvent which can be used for simultaneous estimation of both drugs in a mixture.
Melting point determination is an essential preformulation and physicochemical characterization parameter that supports HPLC method development. Although melting point analysis is not directly involved in chromatographic separation, it provides valuable information regarding the purity, crystallinity, and thermal behaviour of the drug substance, which indirectly influence method development and optimization. Melting point of Atorvastatin (ATOR) & Ezetimibe (EZTB) were determined by placing small amount of sample in capillary tube closed at one end and holds the capillary.
RESULT & DISCUSSION
Now a day’s drugs are commonly used clinically and analyst is required to develop suitable method for their analysis. A fixed dose combination containing Atorvastatin (ATOR) & Ezetimibe (EZET) is recently available in market as tablet dosage form.Percent purity of above mentioned drugs were reported by Supplier Company as follows-
1) Atorvastatin (ATOR)- 99.3 %.
2) Ezetimibe (EZTB) - 99.7 %
Primary this was not analysed in our study and the % purity stated by the suppliers was taken as standard for comparison studies The physico-chemical characterization of drug molecule is important with regard to its purity, identification in development and validation of analytical method. The various tools used for characterization of drug molecules include melting point, UV spectroscopy, solubility study, etc. The solubility study, melting point analysis, UV spectroscopy of the drug was done.
Solubility Study
Atorvastatin
Table No 03:Result of solubility study of Atorvastatin
|
Sr. No |
Solvent / Medium |
Solubility Observation |
|
1 |
Water |
Practically insoluble |
|
2 |
Methanol |
Slightly soluble |
|
3 |
Ethanol |
Sparingly soluble |
|
4 |
Acetonitrile |
Freely soluble |
|
5 |
Dimethylsulfoxide (DMSO) |
Freely soluble |
The solubility characteristics of Ezetimibe significantly influence the development of an effective RP-HPLC method. Ezetimibe is practically insoluble in water and exhibits good solubility in organic solvents such as methanol, ethanol, and chloroform, while being sparingly soluble in acetonitrile. Due to its lipophilic nature and poor aqueous solubility, the use of a higher proportion of organic solvent in the mobile phase is essential to ensure complete dissolution and proper elution of the drug.
Chromatographic conditions:
The following chromatographic conditions were established by trial and error and were kept constant throughout method.
Column : Intersil 4.6 (id) x 250 mm
Particle size packing : 5 µm
Stationary phases : C18 Intersil
Mobile phase : Acetonitrile: Buffer (70:30) pH 3.5
Detection wavelength : 254 nm
Flow rate : 1 ml/min.
Temperature : Ambient
Sample size : 20 uL.
SUMMERY
Analytical method development and validation for the simultaneous estimation of Atorvastatin and Ezetimibe in bulk and it’s dosage form by using RP-HPLCHPLC has gained the valuable position in the field of analysis due to ease of performance, specificity, sensitivity and the analysis of sample of complex nature. This technique is commonly used for the quantitative estimation of the drugs from their formulation as well as for studying their metabolites of drugs and their estimation in their biological fluids. This method offers advantages of estimating the constituents for the multicomponant system without prior separation and even nano quantities can be estimated. This technique was employed in the present investigation for simultaneous estimation of Atorvastatin (ATOR) & Ezetimibe (EZTB) in tablet dosage form. Careful evaluation of various parameters influencing analysis is an important aspect for the development of analytical method. In order to establish RP-HPLC method the following parameters were studied. HPLC with Inert sil 4.6 (id) x 250 mm column and UV detector was used for the study. The standard and sample solution of ATOR and EZTB were prepared in mobile phase. Different pure solvents of varying polarity in different proportions were tried as mobile phase for development of the chromatogram. During selection and optimization of the mobile phase it was observed that the sharpness of the peak is achieved with increasing the proportion of acetonitrile whereas the increased proportion of aqueous resulted in broadening of the peak. The mobile phase that was found to be most suitable was ACN: Buffer (70:30) pH 3.5 and detection wavelength 254 nm was selected for the evaluation of the chromatogram of both drugs. The selection of the wavelength was based on the λ max obtained by scanning of standard laboratory mixture. This system gave good resolution and optimum retention time with appropriate tailing factor (<2). The mean values of system suitability test result are depicted in Table below.
Table No.5 Summary of system suitability test results
|
Sr. No. |
Parameter |
ATOR |
EZTB |
|
1. |
Peakarea |
131074.48 |
111636.03 |
|
2. |
Retentiontime (min) |
3.633 |
6.171 |
|
3. |
Asymmetry |
1.095 |
1.084 |
|
4. |
Efficiency |
102586.18 |
110197.42 |
CONCLUSION
The present study successfully developed and validated a simple, precise, accurate, and robust RP-HPLC method for the simultaneous estimation of Atorvastatin and Ezetimibe in bulk and pharmaceutical dosage forms. Various chromatographic parameters were optimized to achieve efficient separation and reliable results. The mobile phase consisting of acetonitrile and buffer in the ratio of 70:30 (v/v) with pH adjusted to 3.5 was found to be most suitable, providing well-resolved, sharp, and symmetrical peaks for both drugs. Detection was carried out at 254 nm, which offered adequate sensitivity and specificity for the analysis. The method exhibited excellent linearity for both Atorvastatin and Ezetimibe over the selected concentration ranges, with correlation coefficients close to 1. The validation of the method was performed in accordance with ICH guidelines, and all parameters met the acceptable criteria. Accuracy studies showed satisfactory percentage recovery, indicating no interference from excipients. Precision studies demonstrated low %RSD values, confirming the repeatability and reproducibility of the method. The limits of detection (LOD) and quantification (LOQ) indicated that the method is sufficiently sensitive for routine analysis. Robustness studies revealed that small, deliberate variations in chromatographic conditions such as flow rate and pH did not significantly affect the results, confirming the reliability of the method. The developed method was successfully applied to the analysis of pharmaceutical dosage forms, and the assay results were found to be within acceptable limits. Therefore, the proposed RP-HPLC method can be effectively utilized for routine quality control and simultaneous estimation of Atorvastatin and Ezetimibe in bulk and dosage forms. The method is rapid, economical, sensitive, and suitable for routine analytical applications in pharmaceutical industries.
REFERENCES


Sakshi Pinjarkar, Dr. Nusrat Sayyad, Dr. Nitin Bhajipale, A Research on RP-HPLC Method Development & Validation for the Simultaneous Determination of Atorvastatin & Ezetimibe in Bulk & its Pharmaceutical Formulation, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 309-319. https://doi.org/10.5281/zenodo.20491730
 10.5281/zenodo.20491730
10.5281/zenodo.20491730