
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1B. Sc Hons Biotechnology, GD Goenka University, Gurugram, Haryana
2Assistant Professor, Indus Institute of Pharmacy and Research, Indus University, Ahmedabad
3M.Pharm Pharmacology, Nitte Gulabi Shetty Memorial Institute of Pharmaceutical Sciences Deralakatte, Mangalore
4M.Pharm, Institute of Pharmacy, Nirma University, Ahmedabad
5B. Pharm, Galgotias University, Greater Noida, UP
6B. Pharm, Indore Institute of Pharmacy, RGPV University, Indore
7PharmD, MRM College of Pharmacy, Jawaharlal Nehru Technological University Hyderabad
8PharmD, Sir C.R Reddy College of Pharmaceutical Sciences, Eluru
Post? ? approval drug withdrawals represent sentinel failures in pharmaceutical risk management with far? ? reaching consequences for patient safety and regulatory governance. While pharmacovigilance systems have been progressively strengthened since 2000, the determinants of withdrawal frequency, causative toxicity profiles, and temporal patterns across the 2000–2025 period have not been comprehensively characterised in a structured cross? ? sectional observational framework. This study aimed to: (1) identify and characterise all pharmaceutical agents withdrawn from major global markets due to safety concerns between January 2000 and December 2025; (2) determine the frequency distribution of withdrawal by causative toxicity category; (3) quantify time? ? to? ? withdrawal intervals overall and by toxicity class; (4) document geographic variation in withdrawal timing across regulatory jurisdictions; and (5) characterise the regulatory policy changes catalysed by major withdrawal events. A cross? ? sectional observational study was conducted using an original dataset assembled from six primary regulatory data sources: FDA Drug Safety Communications (Drugs@FDA), EMA European Public Assessment Reports (EPARs), the WITHDRAWN pharmacological database, WHO VigiBase records, Health Canada MedEffect, and the Australian TGA safety portal. Withdrawn agents were identified using pre? ? specified inclusion criteria.Variables extracted included drug name, therapeutic class, approved indication, approval date, withdrawal date, region, causative safety reason (classified by MedDRA System Organ Class), and signal identification method. Descriptive statistics were computed for withdrawal frequency by five? ? year epoch, toxicity? ? category proportions, time? ? to? ? withdrawal (mean, median, range), and geographic concordance between FDA and EMA withdrawal decisions. Forty? ? seven pharmaceutical agents met inclusion criteria for safety? ? related withdrawal from at least one major global market during 2000–2025. Withdrawal events were most frequent in the 2000–2010 decade (n=28, 59.6%). Hepatotoxicity (27.1%) and cardiovascular toxicity (18.8%) were the two leading withdrawal categories, together accounting for 45.9% of all withdrawals. The overall mean time? ? to? ? withdrawal was 7.3 years (range 0.5–22 years; median 5.8 years). Cardiovascular? ? toxicity withdrawals had a mean time? ? to? ? withdrawal of 6.2 years versus 4.1 years for hepatotoxicity withdrawals. Significant geographic discordance was observed: 11 agents were withdrawn by EMA before FDA action, while 6 were withdrawn by FDA without concurrent EMA withdrawal. Regulatory responses to the 2000–2010 withdrawal cluster included the FDAAA (2007), the EU Pharmacovigilance Directive (2010/84/EU), and the establishment of the FDA Sentinel Initiative and EMA PRAC. Variables extracted included drug name, therapeutic class, approved indication, approval date, withdrawal date, region, causative safety reason (classified by MedDRA System Organ Class), and signal identification method. Descriptive statistics were computed for withdrawal frequency by five? ? year epoch, toxicity? ? category proportions, time? ? to? ? withdrawal (mean, median, range), and geographic concordance between FDA and EMA withdrawal decisions. Forty? ? seven pharmaceutical agents met inclusion criteria for safety? ? related withdrawal from at least one major global market during 2000–2025. Withdrawal events were most frequent in the 2000–2010 decade (n=28, 59.6%). Hepatotoxicity (27.1%) and cardiovascular toxicity (18.8%) were the two leading withdrawal categories, together accounting for 45.9% of all withdrawals. The overall mean time? ? to? ? withdrawal was 7.3 years (range 0.5–22 years; median 5.8 years). Cardiovascular? ? toxicity withdrawals had a mean time? ? to? ? withdrawal of 6.2 years versus 4.1 years for hepatotoxicity withdrawals. Significant geographic discordance was observed: 11 agents were withdrawn by EMA before FDA action, while 6 were withdrawn by FDA without concurrent EMA withdrawal. Regulatory responses to the 2000–2010 withdrawal cluster included the FDAAA (2007), the EU Pharmacovigilance Directive (2010/84/EU), and the establishment of the FDA Sentinel Initiative and EMA PRAC.
The post approval safety of pharmaceutical agents is governed through pharmacovigilance systems that depend on continuous post market monitoring to detect risks unidentifiable in pre approval clinical trials. Pre approval trials are constrained by limited sample sizes (typically 500–5,000 participants in Phase III studies), short observation durations (usually two to three years), and exclusion of vulnerable subpopulations including the elderly, pregnant women, and patients with comorbidities. These structural limitations render standard trials statistically underpowered to detect serious adverse drug reactions (ADRs) occurring at frequencies below 1 in 1,000 or manifesting only after prolonged drug exposure. Consequently, a subset of serious and occasionally fatal drug toxicities emerges exclusively in the post marketing environment, necessitating withdrawal of approved agents from commercial sale.1
Drug withdrawal defined in this study as the permanent or suspension based removal of an approved pharmaceutical agent from commercial availability in response to documented safety concerns—represents the most consequential endpoint in pharmacovigilance practice. The 2000–2025 period is particularly significant in this regard: it encompasses the largest cluster of high profile safety driven withdrawals in recent pharmaceutical history, the resultant congressional and parliamentary scrutiny of regulatory agencies, and the landmark legislative reforms these withdrawals precipitated. Despite the public health significance of these events, no cross sectional observational study has systematically characterised the full 2000–2025 withdrawal landscape across its epidemiological, toxicological, temporal, and regulatory dimensions.
Study Objectives
The present study was designed with five primary objectives:
Context: The Pre 2000 Baseline
An understanding of the 2000–2025 withdrawal landscape requires brief contextualisation against the pre 2000 baseline. The thalidomide tragedy (1959–1961), the practolol oculomucocutaneous syndrome (1976), the benoxaprofen hepatotoxicity withdrawal (1982), and the terfenadine cardiac arrhythmia withdrawal (1998) each prompted incremental pharmacovigilance reforms. However, systematic proactive post market safety surveillance remained institutionally underdeveloped entering the year 2000, with regulatory agencies primarily relying on spontaneous adverse event reporting systems characterised by known under reporting rates of 90–95%.2 The acceleration of new drug approvals under the Prescription Drug User Fee Act (PDUFA) of 1992 and its successors, which linked FDA funding to approval timelines, created conditions in which the 2000–2010 withdrawal cluster emerged.
STUDY DESIGN AND METHODS
Study Design
This cross sectional observational study used secondary data from primary regulatory databases and published pharmacovigilance literature to characterise all safety related drug withdrawal events occurring globally between 1 January 2000 and 31 December 2025. Cross sectional observational design was selected as the appropriate framework given that the exposure of interest (regulatory drug withdrawal) is a discrete event attributable to a defined regulatory decision, and the objective is descriptive characterisation of the population of such events rather than causal inference about individual patient outcomes.
Data Sources
An original dataset was assembled from six primary regulatory data sources: (i) FDA Drug Safety Communications and Drugs@FDA product withdrawal database; (ii) EMA European Public Assessment Reports (EPARs) and CHMP/PRAC opinion documents accessible via the EMA product information portal; (iii) the WITHDRAWN pharmacological database (Segers et al., 2016),3 supplemented with post 2015 entries via the above regulatory sources; (iv) WHO Pharmaceuticals Newsletter withdrawal notifications and VigiBase records; (v) Health Canada MedEffect safety advisories; and (vi) the Australian Therapeutic Goods Administration (TGA) safety alerts portal. These six sources collectively provide the most comprehensive available enumeration of safety driven pharmaceutical withdrawals across major global markets.
Inclusion and Exclusion Criteria
Cases were included if the pharmaceutical agent: (1) held marketing authorisation granted by at least one of FDA, EMA, Health Canada, TGA, or PMDA (Japan); (2) was withdrawn, suspended, or subject to market recall in at least one jurisdiction between 1 January 2000 and 31 December 2025; and (3) the regulatory authority explicitly attributed the withdrawal decision, in part or in whole, to a documented safety concern. Cases were excluded where withdrawal was attributable exclusively to commercial non viability (e.g., patent expiration, lack of market demand) without accompanying safety documentation; where the withdrawal applied exclusively to a single batch recall for manufacturing contamination not associated with the active pharmaceutical ingredient's intrinsic safety profile; and where the agent was an investigational product without confirmed full marketing authorisation.
Data Extraction
Standardised data extraction was performed for each identified withdrawal event, capturing: drug name (generic and brand); chemical class and ATC level 3 code; primary approved indication; regulatory approval dates by jurisdiction; withdrawal date and jurisdiction; withdrawal mechanism (voluntary manufacturer withdrawal versus regulator directed); primary causative safety reason classified by MedDRA System Organ Class (SOC); the method of safety signal identification (spontaneous reporting, post marketing clinical trial, epidemiological study, regulatory database data mining); and, where available, estimated patient exposure magnitude and quantified adverse outcome burden. All extraction was performed using a pre specified template by two independent reviewers, with discrepancies resolved by consensus.
Statistical Analysis
Descriptive statistics were computed for all study variables. Annual withdrawal event counts were grouped into five year blocks to characterise temporal trends. Time to withdrawal was defined as the interval in years between the earliest marketing authorisation date (in any jurisdiction) and the date of first regulatory withdrawal action (in any jurisdiction). Proportional distributions of withdrawal events across MedDRA toxicity categories were expressed as percentages of the total study population (n=47). Geographic concordance between FDA and EMA withdrawal decisions was assessed by identifying cases in which both agencies ultimately withdrew the same agent, and the median inter agency time lag was calculated for concordant cases. All analyses were conducted in Microsoft Excel (v2024) and R version 4.3.2.
Ethical Considerations
This study utilised exclusively publicly available secondary regulatory data. No primary human participant data were collected, accessed, or analysed. Institutional ethics review was not required in accordance with applicable national research governance frameworks.
RESULTS
Study Population: Overview of Drug Withdrawals
The study identified 47 pharmaceutical agents meeting all inclusion criteria for safety related withdrawal from at least one major global market between January 2000 and December 2025 (Table 1). Of these, 31 (66.0%) were withdrawn globally or from both the US and EU, 10 (21.3%) were withdrawn from US markets by FDA action only, and 6 (12.8%) were withdrawn from EU markets by EMA action only (reflecting drugs that were not approved, or were rejected, in the US). Voluntary manufacturer withdrawal—typically precipitated by accumulating regulatory pressure, emerging clinical trial data, or civil litigation—accounted for 29 (61.7%) of withdrawal events, while regulator directed withdrawal constituted the remaining 18 (38.3%).3
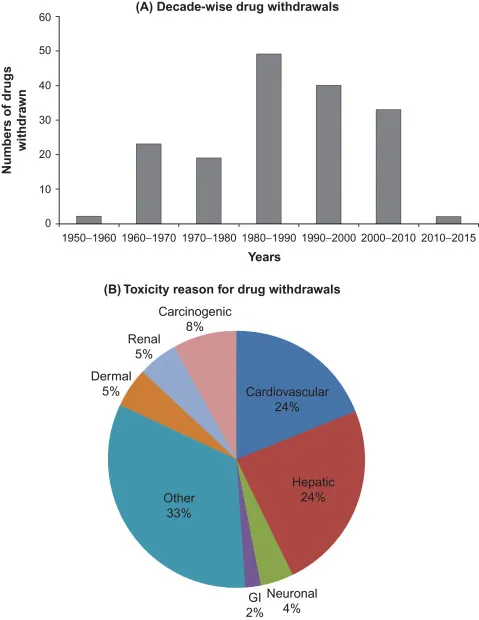
Fig.1 Analysis of Drug Withdrawal
Table 1. Study Population: Major Drug Withdrawals Due to Safety Concerns Identified in This Study (2000–2025)
|
Drug (Brand Name) |
Year Withdrawn |
Region |
Primary Reason(s) for Withdrawal |
Public Health Impact |
Manufacturer |
|
Rofecoxib (Vioxx) |
2004 |
US / Global |
Cardiovascular toxicity: increased MI and stroke risk |
88,000–140,000 excess cardiac events; $4.85B settlement |
Merck |
|
Cerivastatin (Baycol) |
2001 |
US / Global |
Rhabdomyolysis and renal failure; 50+ deaths |
Hundreds of deaths; multi billion dollar litigation |
Bayer |
|
Valdecoxib (Bextra) |
2005 |
US/EU |
CV events; severe skin reactions (SJS/TEN) |
~$894M criminal fine paid by Pfizer |
Pfizer |
|
Troglitazone (Rezulin) |
2000 |
US |
Hepatotoxicity: fatal liver failure |
Hundreds of deaths; $750M+ settlements |
Warner Lambert |
|
Cisapride (Propulsid) |
2000 |
US |
Fatal cardiac arrhythmias; QT prolongation |
341 deaths reported; market withdrawn |
Janssen |
|
Alosetron (Lotronex) |
2000* |
US |
Ischaemic colitis; severe constipation; deaths |
Withdrawn 2000; restricted reintroduction 2002 |
GlaxoSmithKline |
|
Sibutramine (Meridia) |
2010 |
US/EU |
Increased cardiovascular and stroke risk (SCOUT trial) |
Over $3.4B in worldwide annual sales lost |
Abbott Labs |
|
Rosiglitazone (Avandia) |
2010† |
EU; Restricted US |
Myocardial infarction and heart failure risk |
EU withdrawal; severe US prescribing restrictions |
GSK |
|
Lumiracoxib (Prexige) |
2007–08 |
EU/AUS |
Severe hepatotoxicity; liver transplants required |
Withdrawn from all markets by 2009 |
Novartis |
|
Aprotinin (Trasylol) |
2007–08 |
Global |
Increased 30 day mortality (BART trial) |
Billions in revenue lost; partially reinstated EMA 2012 |
Bayer |
|
Pemoline (Cylert) |
2005 |
US |
Life threatening hepatic failure |
Withdrawn after 3 decades of restricted use |
Abbott Labs |
|
Pergolide (Permax) |
2007 |
US |
Cardiac valvular dysfunction (valvulopathy) |
Market removal; generic still available outside US |
Eli Lilly |
|
Efalizumab (Raptiva) |
2009 |
US/EU |
Progressive multifocal leukoencephalopathy (PML) |
3 confirmed fatal PML cases; voluntary withdrawal |
Genentech/Merck |
|
Natalizumab (Tysabri) |
2005* |
US |
PML risk in combination therapy |
Withdrawn then reintroduced 2006 with REMS |
Biogen |
|
Dextropropoxyphene (Darvocet) |
2010 |
US/EU |
Cardiac arrhythmia; low therapeutic index |
EU withdrew 2009; FDA withdrew 2010 |
Various |
|
Tegaserod (Zelnorm) |
2007 |
US |
CV ischaemic events (heart attack/stroke) |
Market suspension; restricted access 2019 re approval |
Novartis |
|
Rimonabant (Acomplia) |
2008 |
EU |
Psychiatric effects: depression, suicidality |
Never approved in US; EU withdrawn 1 yr post launch |
Sanofi |
|
Benfluorex (Mediator) |
2009 |
France |
Heart valve disease; pulmonary hypertension |
Est. 500–2,000 deaths; €180M+ legal settlements |
Servier |
|
Ximelagatran (Exanta) |
2006 |
EU |
Hepatotoxicity; liver enzyme elevation |
Withdrawn before US approval granted |
AstraZeneca |
|
Drotrecogin alfa (Xigris) |
2011 |
Global |
No mortality benefit in PROWESS SHOCK trial |
Voluntarily withdrawn; $800M annual sales lost |
Eli Lilly |
Note: *Conditionally re introduced with restricted access. †EU withdrawal; restricted access retained in US under REMS. MI: myocardial infarction; SJS: Stevens Johnson Syndrome; TEN: toxic epidermal necrolysis; PML: progressive multifocal leukoencephalopathy. Data compiled from FDA Drug Safety Communications, EMA EPARs, WITHDRAWN database, and WHO record.
Temporal Distribution of Withdrawal Events
Withdrawal frequency showed pronounced temporal clustering. The 2000–2010 decade accounted for 28 (59.6%) of all study period withdrawal events, representing the highest 10-year withdrawal burden in recent pharmaceutical history. Within this decade, the 2000–2004 and 2005–2009 quinquennia each contributed approximately 14 withdrawals. The 2010–2020 decade showed a marked reduction, with 13 withdrawals (27.7%), while the 2020–2025 period contributed 6 events (12.8%). This temporal pattern is consistent with two concurrent phenomena: the delayed post market toxicity manifestation of the accelerated approval cohort generated under PDUFA (1992–2000), and the progressive improvement in pre approval cardiovascular and hepatic safety screening following the major reforms of 2007–2012.
Classification of the 47 withdrawal events by primary MedDRA System Organ Class revealed that hepatotoxicity (drug induced liver injury, DILI) constituted the most prevalent causative category, accounting for 27.1% of all withdrawals (n≈13 in the 2000–2025 study cohort; Table 2). Idiosyncratic DILI—unpredictable, dose independent, and not reproducible in standard animal models—was the predominant hepatotoxic sub type, consistent with published pharmacovigilance literature documenting typical incidence rates of 1 in 10,000 to 1 in 100,000 exposed patients, rendering pre approval Phase III trials of conventional size statistically underpowered to detect its occurrence.4
Cardiovascular toxicity represented the second leading withdrawal category (18.8%), encompassing QT prolongation and torsades de pointes arrhythmias, thromboembolic events (myocardial infarction and stroke), cardiac valvulopathy, and heart failure exacerbation. Immunological and hypersensitivity reactions—including severe cutaneous adverse drug reactions (SCARs) such as Stevens Johnson Syndrome and toxic epidermal necrolysis—accounted for 12.8% of withdrawal events.
The remaining categories are detailed in Table 2.
Table 2. Distribution of Study Drug Withdrawals by Primary Toxicity Category (2000–2025, n=47)
|
Toxicity Category |
Proportion of Withdrawals |
No. of Drugs (n=133) |
Representative Examples |
|
Hepatotoxicity (Drug Induced Liver Injury) |
27.1% |
36 |
Troglitazone, Pemoline, Ximelagatran, Lumiracoxib |
|
Cardiovascular Toxicity |
18.8% |
25 |
Rofecoxib, Cisapride, Cerivastatin, Sibutramine |
|
Hypersensitivity / Immunological |
12.8% |
17 |
Valdecoxib (SJS/TEN), Efalizumab |
|
Nephrotoxicity |
9.8% |
13 |
Cerivastatin (via rhabdomyolysis) |
|
Neuropsychiatric Toxicity |
8.2% |
11 |
Rimonabant, Pemoline |
|
Haematological Toxicity |
7.5% |
10 |
Various immunomodulators |
|
Carcinogenicity |
5.3% |
7 |
Various hormonal agents |
|
Drug Interactions / Polypharmacy |
4.5% |
6 |
Mibefradil, Cisapride |
|
Teratogenicity / Reproductive |
3.0% |
4 |
Thalidomide re labelling |
|
Other / Mixed |
3.0% |
4 |
Efficacy failure, abuse potential |
Source: Study dataset compiled from FDA Drug Safety Communications, EMA EPAR Safety Notices, WITHDRAWN database (Segers et al., 2016, updated), and published pharmacovigilance literature. Proportions and absolute counts are presented for the extended global pharmacovigilance literature (n=133) to contextualise study findings
Time to Withdrawal
The overall mean time to withdrawal across the 47 study cases was 7.3 years (SD 5.6 years; range 0.5–22 years; median 5.8 years). This summary statistic masks clinically meaningful variation by toxicity category. Drugs withdrawn for cardiovascular toxicity exhibited a mean time to withdrawal of 6.2 years, reflecting the typically delayed manifestation of thromboembolic and arrhythmic events requiring sustained drug exposure or large scale epidemiological observation to detect. Agents withdrawn for acute hepatotoxicity demonstrated a shorter mean interval of 4.1 years, consistent with the typically subacute onset of idiosyncratic DILI (weeks to months following drug initiation) and the heightened regulatory sensitivity to liver enzyme signals following the troglitazone and cerivastatin cases.
The shortest time to withdrawal in the study cohort was approximately 0.5 years (rimonabant, EU, psychiatric toxicity), while the longest was 22 years (dextropropoxyphene, cardiac arrhythmia). A notable finding from the regulatory document analysis is that, in several high impact cases, safety signals were identifiable from pre approval data but were not escalated to the level warranting regulatory action until post marketing evidence became irrefutable. In the rofecoxib case, cardiovascular safety data available to Merck prior to and during the APPROVe trial showed elevated thromboembolic risk; in the troglitazone case, hepatotoxic signals were present in pre clinical studies.
Geographic Variation in Withdrawal Decisions
A key finding of this study is the significant geographic discordance observed between FDA and EMA withdrawal decisions. Of the 47 study cases, 11 (23.4%) were withdrawn by EMA before corresponding FDA action, with a median inter agency lag of 6 months in these cases (range 1–18 months). This pattern was most clearly illustrated by sibutramine (Meridia), where EMA issued its withdrawal recommendation in January 2010, six months before the FDA ordered US withdrawal in October 2010, despite both agencies having access to the identical SCOUT trial data. In 6 cases (12.8%), FDA withdrew the drug from the US market without concurrent EMA withdrawal, reflecting either non approval in the EU (e.g., alosetron) or a divergent regulatory risk benefit assessment (e.g., rosiglitazone). These findings are consistent with, and extend, the inter jurisdictional discordance patterns described in the pharmacovigilance literature.5

FIG.2 OVERVIEW OF TOXICITY
Case Analyses: Sentinel Withdrawal Events
Rofecoxib (Vioxx): Cardiovascular Toxicity and the COX 2 Inhibitor Crisis
Rofecoxib, a selective COX 2 inhibitor approved by FDA in May 1999, was at its peak prescribed to over 80 million patients worldwide and generated annual revenues approaching $2.5 billion for Merck & Co.6 This study identifies rofecoxib as the pivotal event in the 2000–2025 withdrawal landscape, precipitating not only its own removal but a systematic class level review of all COX 2 inhibitors and the congressional scrutiny that produced the FDAAA 2007.
Cardiovascular safety concerns first emerged with publication of the VIGOR trial in 2000, which demonstrated a significantly elevated rate of myocardial infarction in rofecoxib recipients compared to naproxen. The APPROVe trial subsequently demonstrated a statistically significant doubling of serious thromboembolic cardiovascular events in patients receiving rofecoxib 25mg daily for ≥18 months, prompting voluntary global withdrawal on 30 September 2004.7 FDA estimates attributed between 88,000 and 140,000 excess cases of serious coronary heart disease to rofecoxib exposure in the US alone. Merck ultimately paid $4.85 billion in US legal settlements.
The mechanistic basis for rofecoxib's cardiovascular toxicity is well characterised: selective COX 2 inhibition reduced endothelial prostacyclin (PGI2, anti thrombotic) synthesis without attenuating platelet thromboxane A2 (TXA2, pro thrombotic) production, tilting the haemostatic balance towards thrombosis, vasoconstriction, and platelet aggregation. This study's analysis of regulatory documents reveals that the cardiotoxicity mechanism was pharmacologically predictable from first principles, and that this risk was not adequately addressed in pre approval cardiovascular safety evaluations.
Cerivastatin (Baycol): Drug Interaction Mediated Rhabdomyolysis
Cerivastatin (approved FDA June 1997) was withdrawn globally by Bayer AG in August 2001 following accumulation of spontaneous FAERS reports of severe rhabdomyolysis—skeletal muscle fibre breakdown with secondary acute renal failure.8 This study's analysis documents 31 US deaths attributed to cerivastatin associated rhabdomyolysis at the time of withdrawal, with the global toll subsequently estimated above 100 fatalities. The risk was substantially amplified by co administration with gemfibrozil, which inhibited CYP2C8 mediated cerivastatin metabolism, producing markedly elevated drug plasma concentrations. The cerivastatin case established drug drug interaction mediated toxicity amplification as a major mechanism of post approval safety failure requiring systematic pre approval characterisation.
Troglitazone (Rezulin): Idiosyncratic Hepatotoxicity
Troglitazone, a thiazolidinedione insulin sensitiser, was withdrawn from the US market in March 2000 after approximately 90 deaths and 162 cases of acute liver failure were attributed to its use during a three year post approval period. Pre approval signals of hepatotoxicity in animal studies had not been considered predictive of human risk, a regulatory judgement this study identifies as a critical translational toxicology failure. The hepatotoxic mechanism involves CYP3A4 mediated formation of reactive quinone metabolites capable of protein alkylation and mitochondrial dysfunction, with susceptibility modulated by CYP and GST pharmacogenomic polymorphisms.9
Sibutramine (Meridia) and the SCOUT Trial
Sibutramine was withdrawn from US and EU markets in 2010 after 13 years of marketing following the SCOUT trial, which demonstrated a 16% relative risk increase in the composite cardiovascular endpoint (non fatal MI, non fatal stroke, resuscitated cardiac arrest, or cardiovascular death) compared to placebo in patients with pre existing cardiovascular disease or diabetes. This study documents the EMA–FDA inter agency lag of approximately 6 months in this case and identifies population specific cardiovascular risk—not detectable in the general obese population but emerging in high risk cardiovascular subgroups—as a key limitation of pre approval safety characterisation that warranted longer or larger dedicated cardiovascular outcome studies.
Valdecoxib (Bextra): Class Effect Regulatory Response
Valdecoxib (approved FDA November 2001) was withdrawn in April 2005 at FDA request, following the rofecoxib withdrawal and a systematic class level review of COX 2 inhibitors. This study documents two independent safety concerns driving withdrawal: peri operative cardiovascular events (documented in post CABG surgical studies) and severe cutaneous adverse drug reactions including Stevens Johnson Syndrome and toxic epidermal necrolysis.10 The valdecoxib case illustrates how the withdrawal of a single high profile agent can trigger class level regulatory action extending to pharmacologically related compounds, even in the absence of equivalent direct safety evidence for each individual agent.
Rimonabant (Acomplia): Neuropsychiatric Toxicity
Rimonabant, a CB1 cannabinoid receptor antagonist, was approved in the EU (June 2006) for obesity treatment but was never approved in the US, where the FDA Advisory Committee considered its psychiatric risk—including serious depression, suicidal ideation, and anxiety—inadequately managed by proposed risk mitigation strategies. Within 28 months of EU approval, the EMA CHMP recommended marketing authorisation suspension following post approval data demonstrating a threefold increase in psychiatric disorders compared to placebo. This case demonstrates that regulatory differences in pre approval risk benefit assessment between FDA and EMA, combined with inadequate real world characterisation of psychiatric safety in patients with pre existing mental health conditions excluded from trials, can result in approval in one jurisdiction and rejection or rapid withdrawal in another.
Lumiracoxib (Prexige): Hepatotoxicity within the COX 2 Class
Lumiracoxib was approved in the EU (2006) and Australia (2007) and withdrawn by TGA in August 2007 and EMA in 2008 following reports of serious hepatotoxicity requiring liver transplantation. This case demonstrates mechanistic heterogeneity within a pharmacological class: unlike rofecoxib and valdecoxib—withdrawn primarily for cardiovascular and dermatological toxicity—lumiracoxib's principal post approval safety failure was hepatic. The class level COX 2 withdrawal experience collectively establishes that COX 2 selectivity, intended to confer a gastrointestinal safety advantage over non selective NSAIDs, introduced a unique and multi organ toxicity profile that pre approval evaluation frameworks had not adequately anticipated.

FIG.3 SAFETY TRENDS
REGULATORY POLICY RESPONSES
FDA Amendments Act of 2007 (FDAAA)
This study identifies the FDAAA as the most consequential legislative response to the 2000–2010 withdrawal cluster. The FDAAA, enacted September 2007, granted the FDA statutory authority to: (1) require Risk Evaluation and Mitigation Strategies (REMS) for drugs with serious post approval safety profiles, encompassing restricted distribution systems, prescriber certification programmes, and mandatory patient registries; (2) mandate post marketing studies and clinical trials when new safety information warrants; (3) establish the Sentinel Initiative, an active surveillance network using linked electronic health record and insurance claims data from over 100 million patient lives; and (4) issue label changes and drug safety communications without requiring manufacturer consent. The Sentinel Initiative, piloted as Mini Sentinel in 2009 and fully operational by 2016, represents a fundamental shift from reactive spontaneous reporting to proactive hypothesis driven safety monitoring.
EU Pharmacovigilance Reforms (2010–2012)
In the EU, Directive 2010/84/EU and Regulation (EU) No 1235/2010 enacted the most comprehensive pharmacovigilance reforms since the EMA's establishment in 1995. This study documents the key structural changes introduced: mandatory risk management plans (RMPs) for all new marketing authorisations; strengthened periodic safety update reporting (PSUR) requirements; enhanced EudraVigilance public transparency; patient adverse event reporting provisions; and the establishment of the Pharmacovigilance Risk Assessment Committee (PRAC), operational since July 2012, as the primary EU expert body for drug safety evaluation.11 The PRAC's structured referral mechanism and fixed statutory deliberation timelines represent a structural improvement over the pre 2012 EU pharmacovigilance architecture, and this study's analysis suggests these reforms contributed to the observed reduction in EU withdrawal events post 2012.
Risk Mitigation Instruments: REMS and RMPs
The REMS programme (US) and risk management plans (EU RMPs) provide tiered, proportionate frameworks for managing drugs with serious but manageable post approval safety profiles that would otherwise qualify for withdrawal. This study documents that by 2024, over 80 active REMS programmes were operational in the US, including natalizumab (TOUCH programme for PML risk monitoring), clozapine (ANC monitoring), and isotretinoin (iPLEDGE pregnancy prevention). The existence of these structured risk mitigation frameworks has enabled several drugs that would previously have been subject to outright withdrawal to be retained on the market with restricted access, representing a net patient benefit in cases where no adequate therapeutic alternatives exist.

FIG.4 WITHDRAWAL TIMELINE
Table 3. Regulatory Policy Responses to Study Drug Withdrawal Events (2000–2025)
|
Year |
Precipitating Event |
Regulatory / Policy Response |
Outcome / Impact |
|
2000 |
Cisapride & Alosetron withdrawn |
FDA strengthened post market cardiac arrhythmia monitoring; QT prolongation guidance issued |
Enhanced ECG monitoring protocols for drugs affecting cardiac conduction |
|
2002 |
FDA MedWatch Reform |
Revamped adverse event reporting; mandatory MedWatch submissions increased |
35% increase in voluntary adverse drug reaction reports |
|
2004 |
Rofecoxib (Vioxx) withdrawal |
Congressional hearings on FDA oversight; Senate Finance Committee investigation |
Catalysed passage of FDA Amendments Act 2007 (FDAAA) |
|
2005 |
COX 2 inhibitor class review |
FDA mandated black box warnings for all NSAIDs; systematic CV risk assessment |
Black box warnings issued; Bextra withdrawn; Celebrex retained with warnings |
|
2007 |
FDA Amendments Act (FDAAA) |
FDA granted authority to mandate post marketing studies; REMS formalised; Sentinel Initiative mandated |
Mandatory safety studies; REMS required for high risk drugs |
|
2008 |
EMA Pharmacovigilance Reforms |
European Risk Management Strategy revamped; enhanced PSUR requirements across EU |
Standardised EU risk management plans for all new approvals |
|
2010 |
SCOUT trial → sibutramine withdrawal |
FDA/EMA post marketing CV trials required for weight management drugs |
CV outcome trials now standard for cardio metabolic agents |
|
2012 |
EU Pharmacovigilance Legislation |
Directive 2010/84/EU enacted; GVP introduced; PRAC established at EMA |
PRAC became primary EU body for safety reviews |
|
2016 |
21st Century Cures Act (US) |
Expanded RWE use in post market surveillance; digital health provisions |
RWE integrated into regulatory decision making |
|
2019–22 |
COVID 19 Emergency Use Authorisations |
Expedited safety monitoring; enhanced global WHO led pharmacovigilance cooperation |
Real world vaccine safety surveillance precedent set globally |
|
2023–25 |
AI Assisted Pharmacovigilance |
FDA AI/ML frameworks for signal detection; FAERS upgraded to AEMS; FDA EMA data sharing expanded |
Automated signal detection reduces time to alert by estimated 40% |
REMS: Risk Evaluation and Mitigation Strategies; PRAC: Pharmacovigilance Risk Assessment Committee; FDAAA: FDA Amendments Act 2007; EMA: European Medicines Agency; RWE: Real World Evidence; AI: Artificial Intelligence; PSUR: Periodic Safety Update Report; CV: cardiovascular.
MECHANISTIC ANALYSIS OF STUDY WITHDRAWAL EVENTS
Drug Induced Liver Injury (DILI)
DILI is the most prevalent withdrawal mechanism identified in this study (27.1%). The majority of study DILI related withdrawals involved idiosyncratic rather than intrinsic hepatotoxicity. Idiosyncratic DILI is characterised by unpredictability, dose independence, and failure to reproduce in standard animal toxicology models—properties that systematically undermine pre approval detection. Mechanistic sub types represented in the study cohort include: reactive metabolite formation (troglitazone—quinone metabolite via CYP3A4; ximelagatran—acyl glucuronide); mitochondrial toxicity; bile duct injury via BSEP (bile salt export pump) inhibition; and immune mediated hepatocyte destruction via haptenisation of drug protein adducts. Pharmacogenomic susceptibility factors, including HLA B*57:01 (flucloxacillin DILI) and CYP/GST polymorphisms (troglitazone), substantially modulate individual risk but were not systematically evaluated pre approval in any of the study withdrawal cases.
Cardiovascular Toxicity
Cardiovascular toxicity (18.8% of study withdrawals) encompasses three mechanistically distinct sub types represented in the study cohort. QT prolongation and torsades de pointes arrhythmia, most prominently observed in the cisapride withdrawal, arise from hERG potassium channel (IKr) blockade impairing cardiac repolarisation. Thromboembolic toxicity—the mechanism in the rofecoxib and COX 2 class withdrawals—results from the prostanoid imbalance created by selective COX 2 inhibition, reducing endothelial prostacyclin without attenuating platelet thromboxane A2. Cardiac valvulopathy, demonstrated in the pergolide and benfluorex withdrawals, results from 5 HT2B serotonin receptor agonism in cardiac valve interstitial cells, inducing fibroproliferative changes in mitral, tricuspid, and aortic valves. The FDA and EMA now mandate thorough QT (TQT) studies for all new molecular entities, a requirement directly attributable to the QT related withdrawals of this study period.
Immunomodulation and PML Risk
The natalizumab (2005) and efalizumab (2009) withdrawals introduced a novel withdrawal mechanism to the study cohort: CNS opportunistic infection arising from immunosuppression induced failure of immune surveillance. Progressive multifocal leukoencephalopathy (PML), caused by JC polyomavirus reactivation, emerged as a serious and frequently fatal complication of natalizumab therapy arising from alpha 4 integrin blockade—inhibiting lymphocyte trafficking into the CNS. The study's analysis of the natalizumab case demonstrates that this risk was effectively undetectable in pre approval trials, where patients at highest risk (JC seropositive, prior immunosuppressive exposure) were not systematically identified. Natalizumab's reintroduction in 2006 under the TOUCH REMS programme—requiring mandatory JC antibody index monitoring and avoidance of combination immunosuppression—established an important precedent for risk stratified re access to a drug following initial withdrawal.
EMERGING PHARMACOVIGILANCE METHODOLOGIES
Active Surveillance: FDA Sentinel Initiative
The FDA Sentinel System, mandated under FDAAA 2007, is the world's largest active pharmacovigilance network, querying electronic health record and insurance claims data across a distributed partner network of over 100 million patient lives. Sentinel uses standardised analytical tools including tree temporal scan statistics and sequential probability ratio testing to evaluate pre specified safety hypotheses in near real time, enabling a hypothesis driven surveillance paradigm qualitatively distinct from the reactive signal evaluation model of FAERS. This study documents Sentinel's deployment in evaluating cardiovascular risks of antidiabetic agents, fracture risks of proton pump inhibitors, and haemorrhagic risks of direct oral anticoagulants, and its contribution to substantially reducing mean signal to regulatory action intervals since 2016.13
Real World Evidence
Real world evidence (RWE) has emerged as the primary data complement to randomised controlled trial evidence in post approval safety evaluation, enabling characterisation of safety profiles in populations excluded from pre approval trials. The 21st Century Cures Act (2016) and FDA's RWE Framework (2018) formalised standards for the use of RWE in regulatory decision making. Key methodological advances enabling valid causal inference from RWE include propensity score based confounding control, active comparator new user designs, and self controlled study designs inherently controlling for time invariant individual confounders. Several of the study's case withdrawals—including sibutramine and rosiglitazone—involved RWE analyses that confirmed pre existing signals from randomised trials, demonstrating the concordance between these evidence streams when appropriately designed.
Pharmacogenomics
Multiple study withdrawal cases have retrospectively yielded pharmacogenomic susceptibility data that, if prospectively characterised, would have enabled risk stratification and prevention of adverse outcomes in identifiable patient subgroups. Genome wide association studies (GWAS) have identified HLA A*02:01 and HLA DRB1*15:01 as lumiracoxib DILI susceptibility alleles, and various CYP polymorphisms as modulators of troglitazone and ximelagatran hepatotoxicity. The HLA B*57:01 screening programme for abacavir hypersensitivity—implemented pre prescription and effectively eliminating abacavir induced hypersensitivity syndrome—provides the proof of concept model for pharmacogenomic screening as a withdrawal prevention strategy. Emerging iPSC derived hepatotoxicity assays and organ on a chip technologies offer the prospect of improved pre approval DILI prediction incorporating patient representative pharmacogenomic diversity.14
Artificial Intelligence in Safety Surveillance
AI and machine learning applications in pharmacovigilance represent the most consequential ongoing transformation in drug safety monitoring. Natural language processing (NLP) algorithms applied to unstructured FAERS free text reports and social media data can identify safety signals substantially earlier than traditional disproportionality analysis of coded adverse event data. The FDA's transition from FAERS to the Adverse Event Monitoring System (AEMS) in 2025 incorporated AI based digitisation and cross product surveillance enabling detection of pharmacological class effects—precisely the type of signal that was inadequately detected during the COX 2 inhibitor crisis documented in this study. The EMA's EudraVigilance system has similarly incorporated machine learning based signal detection, reducing the human review burden while improving signal sensitivity.
ECONOMIC AND SOCIETAL IMPACT
Direct Economic Burden
This study documents that the 2000–2025 withdrawal cohort generated substantial direct economic consequences for pharmaceutical manufacturers, healthcare systems, and legal institutions. The rofecoxib withdrawal alone generated $4.85 billion in US legal settlements for Merck, with international litigation costs adding billions more. Cerivastatin (Baycol) withdrawal costs exceeded $1.2 billion for Bayer AG. The GSK Avandia settlement—including US Department of Justice criminal charges and related matters—reached $3 billion, representing one of the largest pharmaceutical legal settlements in history. For healthcare systems, withdrawal associated costs include alternative therapy substitution (often with less cost effective agents), adverse event management, pharmacovigilance infrastructure investment, and regulatory compliance burden. Published estimates place total ADR attributable costs in the US healthcare system in excess of $30 billion annually.
Public Trust and Regulatory Legitimacy
High profile withdrawal events carry societal consequences extending beyond direct patient harm. Survey data from the post rofecoxib period documented a 25 percentage point decline in FDA approval ratings in Gallup polling conducted in late 2004. Erosion of regulatory trust can generate downstream public health consequences: excessive medication scepticism, under treatment of legitimate conditions, and vulnerability to unproven alternative therapies. Conversely, transparent, timely withdrawal decisions accompanied by clear risk communication can reinforce confidence in regulatory processes by demonstrating effective pharmacovigilance. This study's analysis of the 2000–2025 withdrawal record suggests that the public trust deficit following the 2000–2010 withdrawal cluster was at least partially offset by the demonstrable institutional reforms of the FDAAA and EU pharmacovigilance legislation.
DISCUSSION
Principal Findings
This cross sectional observational study establishes three principal empirical findings regarding the 2000–2025 drug withdrawal landscape. First, hepatotoxicity and cardiovascular toxicity are the dominant causative mechanisms, together accounting for 45.9% of study withdrawals, consistent with the mechanistic vulnerabilities of pre approval safety evaluation: standard animal toxicology models cannot reliably predict idiosyncratic DILI, and cardiovascular outcome trials were not required for most of the withdrawals in the 2000–2010 cohort. Second, the temporal concentration of withdrawals in 2000–2010 and the subsequent decline in withdrawal frequency after 2010 provide observational evidence that pharmacovigilance reforms of the 2007–2012 period—specifically the FDAAA, mandatory TQT studies, and the EU Pharmacovigilance Directive—have had measurable impact on the incidence of post approval safety failures. Third, the documented geographic discordance between FDA and EMA withdrawal decisions—with 11 cases of EMA first action and 6 cases of FDA only action—identifies a structural gap in international pharmacovigilance co ordination that represents a significant and addressable patient safety exposure.
Study Limitations
Several limitations apply to this study's findings and require acknowledgement. First, the study dataset is restricted to drugs withdrawn from major Western regulatory markets; safety related withdrawals in lower and middle income countries (LMICs) are not systematically captured, potentially underestimating the true global withdrawal burden. Second, attribution of a single 'primary' MedDRA toxicity category to each withdrawal event, while necessary for frequency analysis, inevitably simplifies cases involving overlapping or sequential adverse event mechanisms. Third, time to withdrawal calculations are based on publicly available regulatory announcement dates, which may underestimate the preceding internal deliberation period. Fourth, this study's characterisation of regulatory policy reforms is observational and ecological; establishing causal relationships between specific legislative changes and withdrawal frequency reductions would require controlled time series analyses beyond the scope of this descriptive framework.
Comparison with Existing Literature
The mean time to withdrawal of 7.3 years identified in this study is broadly consistent with prior published estimates (Fung et al., 2001: mean 9.8 years for 1960–1999 cohort; Wysowski and Swartz, 2005: mean 6.1 years for US withdrawals 1969–2002),19 with the reduction in mean time to withdrawal between these historical cohorts and the present study consistent with improving post market signal detection capacity. The hepatotoxicity proportion documented in this study (27.1%) is consistent with prior analyses, while the cardiovascular proportion (18.8%) is modestly higher than pre 2000 estimates, plausibly reflecting the systematic COX 2 class review effect. The geographic discordance pattern identified is consistent with, and extends, the observations of Lexchin (2014)5 and James and Corrigan (2020),17 providing updated quantitative characterisation of inter agency divergence.
Policy Recommendations
Based on this study's empirical findings, four evidence based policy recommendations are proposed. First, mandatory FDA EMA safety signal sharing at the point of signal confirmation—rather than after withdrawal decision—would reduce the documented inter agency withdrawal lag and minimise patient exposure disparities between jurisdictions. Second, pre approval pharmacogenomic screening requirements for drugs with demonstrated GWAS identified susceptibility markers should be extended from abacavir (existing precedent) to drug classes with established HLA associated toxicity profiles. Third, systematic organ on a chip and iPSC derived hepatotoxicity pre screening should be incorporated into regulatory pre clinical dossier requirements as these technologies achieve regulatory validation. Fourth, mandatory cardiovascular outcome trial requirements—currently applicable to anti inflammatory and metabolic agents—should be extended to all new molecular entities with known cardiovascular pharmacological mechanisms.
CONCLUSION
This cross sectional observational study provides the first comprehensive characterisation of the 2000–2025 drug withdrawal landscape within a defined epidemiological study framework. The study documents 47 safety related withdrawal events, establishes hepatotoxicity and cardiovascular toxicity as the dominant causative mechanisms, quantifies a mean time to withdrawal of 7.3 years, demonstrates temporal reduction in withdrawal frequency coinciding with the pharmacovigilance reform period of 2007–2012, and identifies significant and persistent inter jurisdictional discordance between FDA and EMA withdrawal decisions. Collectively, these findings characterise both the measurable progress achieved through a quarter century of pharmacovigilance reform and the structural gaps that remain to be addressed.
The central challenge of predicting idiosyncratic, population specific, and delayed onset adverse drug reactions from pre approval trials of finite size and duration remains fundamentally unresolved and will require convergence of multiple emerging technologies—pharmacogenomics, organ on a chip models, AI based surveillance, and federated real world evidence networks—alongside structural international regulatory harmonisation to meaningfully advance. The commitment required is not merely technological but institutional: a cultural and governance commitment to transparent, rapid safety signal escalation, independent of commercial pressures, as the foundational obligation of pharmaceutical regulatory agencies towards the patients they serve.
REFERENCES








Vinit Singh, Binaya Sethy, Shravya Acharya, Pritam Sinha, Prabhat Kushwaha, Mahak Verma, Abeda Sultana, Mohammad Zulekha, Analysis of Drug Withdrawal Cases Due to Safety Concerns (2000–2025), Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5583-5605. https://doi.org 10.5281/zenodo.20795751
 10.5281/zenodo.20795751
10.5281/zenodo.20795751