
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Regulatory Affairs, Dayanand Education Society’s Dayanand college of pharmacy, Latur, India
Medical device regulation is essential to guaranteeing patient safety, high-quality products, and efficient healthcare delivery. Both the European Union (EU) and India have implemented major changes in regulations in recent years in an effort to improve transparency, bolster oversight, and conform with international norms. The recent changes to medical device regulations in the EU and India can be compared in this study, with emphasis on how they affect compliance requirements and regulatory processes. The Medical Devices Rules, 2017 have been amended in India in a number of ways, such as increasing the number of devices that are notified, introducing voluntary to mandatory licensing transitions, strengthening post-market surveillance under the Central Drugs Standard Control Organization (CDSCO), and improving registration requirements. These modifications are intended to improve patient safety and ease of doing business while establishing a more organized and thorough regulatory framework. In contrast, the previous Medical Device Directive (MDD) was replaced by Regulation (EU) 2017/745 (EU Medical Device Regulation-MDR). Stricter guidelines for clinical evaluation, Unique Device Identification (UDI), heightened scrutiny by Notified Bodies, improved transparency via EUDAMED, and strong post-market surveillance and vigilance systems are all introduced by the MDR. This study assesses and contrasts important regulatory areas impacted by these recent changes, such as post-market obligations, quality management systems, device classification, conformity assessment processes, and clinical evidence requirements. The EU MDR exhibits a more rigorous and developed regulatory framework, especially in clinical data and lifecycle monitoring, even though both jurisdictions place a strong emphasis on a risk-based approach and patient safety. India's developing system has made great strides toward conformity with global norms like ISO 13485 and IMDRF guidelines. The results show that there are still significant disparities in procedural complexity, documentation requirements, and approval timelines despite the growing regulatory convergence. To promote innovation, simplify market access, and guarantee the availability of safe and efficient medical devices globally, regulatory harmonization, the adoption of international best practices, and the fortification of regulatory infrastructure are crucial.
1.What is Medical Device?
A medical device is a piece of equipment used on patients for surgery, diagnosis, or treatment. Hospital beds, contact lenses, hip implants, pacemakers, in-vitro diagnostic equipment, and even simple wooden tongue depressors are all considered medical devices.
According to the WHO, medical devices and medical equipment are not the same thing. It defines a medical device as one that is meant to be used for disease prevention, diagnosis, or treatment, as well as for the detection, measurement, restoration, correction, or modification of the body's structure or function to improve health. Medical equipment does not include single-use, disposable, or implantable devices.
The Food and Drug Administration, Health Canada, the Central Drugs Standard Control Organization (CDSCO) in India, the European Medicines Agency (EMA) in Europe, the Ministry of Health, Labour & Welfare (MHLW) in Japan, and many other organizations have adopted similar definitions with only minor changes.(1)
According to the Europe - A medical device is defined as "any instrument, apparatus, appliance, material, or other product, whether used alone or in combination, including the software necessary for its correct application intended by the manufacturer to be used for human beings for the purpose of:
mechanisms to carry out its primary intended activity in or on the human body but which may be aided in its function by such mechanisms”.
According to India- The medications used for in vitro diagnosis, surgical instruments, blood and blood component collection bags (with or without anticoagulant), mechanical contraceptives, disinfectants, insecticides, and other devices that have been properly registered are all listed in India.(2).
A medical device is any tool, apparatus, software, material, or item that is used either separately or in combination. This includes programming required for the device's authorized use that the manufacturer intended to be used only for symptomatic or possibly curative purposes. The device's inventor thinks that in addition to examination, diagnosis, and relief, people will use it for illness detection, treatment, and prognosis.(3)
Classification:
India- The Indian classification system, which is based on variables like intended use, risk level, mode of distribution, and degree of invasion into the human body, is based on the MDR 2017 Guidelines. The Indian regulatory system is currently undergoing change, and only a small number of product categories require import permits. Any additional medical devices may be voluntarily submitted during this grace period. After this voluntary period, non-regulatory medical equipment of all classes A and B will have 12 months (i.e., by October 1, 2022) to obtain an import license. Class C and D devices have until October 1, 2023, or 24 months, to adhere to the same standard. Medical devices are typically categorized according to the risks they pose; the specific classification of a medical device is determined by its intended use and purpose. Cannulas and stents are just two examples of the many devices that fall under the more specific categories of the CDSCO classification for medical devices.[4-5]
Table no1: Classification of Medical Devices in India (3)
|
Sr. No |
Class |
Risk Level |
Examples |
|
1 |
A |
Low risk |
Surgical Dressing, Surgical gloves, Tongue depressors, and Non-invasive devices |
|
2 |
B |
Low moderate risk |
Catheters, Nebulizers, Blood pressure monitors, and Thermometers |
|
3 |
C |
Moderate high risk |
Catheters, Bone fixation plates, and Surgical drapes |
|
4 |
D |
High risk |
Implantable devices, Heart valves, and Certain active devices |
Table no2: List of Newly Notified Medical Devices (3)
|
Sr. No |
Name of the Device |
Effective from |
|
1 |
X-Ray Machines |
April 1, 2021 |
|
2 |
CT Scan Equipment |
April 1, 2021 |
|
3 |
MRI Equipment |
April 1, 2021 |
|
4 |
PET Equipment |
April 1, 2021 |
|
5 |
Defibrillators |
April 1, 2021 |
|
6 |
Dialysis Machines |
April 1, 2021 |
|
7 |
Bone Marrow Cell Separators |
April 1, 2021 |
|
8 |
All Implantable Medical Devices |
April 1, 2021 |
|
9 |
Ultrasound Devices |
November 1, 2021 |
|
10 |
Disinfectants and Insecticides |
- |
|
11 |
Class A (Measuring or Sterile) and All Class B |
October 1, 2022 |
Table no 3: List of Notified Medical Devices in India (3)
|
Sr. No |
Name of the Device |
Notification Number |
Date of Notification |
|
1 |
Disposable Hypodermic Syringes |
GSR 365 (E) |
17-03-1989 |
|
2 |
Disposable Hypodermic Needles |
GSR 365 (E) |
17-03-1989 |
|
3 |
Disposable Perfusion Sets |
GSR 365 (E) |
17-03-1989 |
|
4 |
In vitro Diagnostic Devices for HIV, HBsAg and HCV |
GSR 601 (E) |
27-08-2002 |
|
5 |
Cardiac Stents |
S.O. 1468 (E) |
06-10-2005 |
|
6 |
Drug Eluting Stents |
S.O. 1468 (E) |
06-10-2005 |
|
7 |
Catheters |
S.O. 1468 (E) |
06-10-2005 |
|
8 |
Intra Ocular Lenses |
S.O. 1468 (E) |
06-10-2005 |
|
9 |
I.V. Cannulae |
S.O. 1468 (E) |
06-10-2005 |
|
10 |
Bone Cements |
S.O. 1468 (E) |
06-10-2005 |
|
11 |
Heart Valves |
S.O. 1468 (E) |
06-10-2005 |
|
12 |
Scalp Vein Set |
S.O. 1468 (E) |
06-10-2005 |
|
13 |
Orthopedic Implants |
S.O. 1468 (E) |
06-10-2005 |
|
14 |
Internal Prosthetic Replacements |
S.O. 1468 (E) |
06-10-2005 |
|
15 |
Ablation Devices |
S.O. 237 (E) |
25-01-2016 |
Table no 4: Medical Device Classification Based on Risk Level in EU(45,46)
|
Sr. No |
Class |
Risk Level |
Examples |
|
1 |
Class I |
Low risk |
Bedpans, Sterile dressings, Gloves, Hospital Beds |
|
2 |
Class IIa |
Medium risk |
Surgical blades, |
|
3 |
Class IIb |
Medium to high risk |
Infusion pumps, |
|
4 |
Class III |
High risk |
Replacement heart valves, Breast implants, Drug‑eluting cardiac stents, Pacemakers, |
METHODOLOGY:
This study analyzes recent changes to medical device regulations in the EU and India using a qualitative, comparative method. Updated regulatory frameworks, official guidelines, policy documents, and peer-reviewed literature were all systematically reviewed. The Central Drugs Standard Control Organization's (CDSCO) revised Medical Devices Rules, 2017 and Regulation (EU) 2017/745 (Medical Device Regulation, MDR) are important sources. Device classification, pre-market approval pathways, clinical evaluation requirements, conformity assessment procedures, quality management systems (QMS), post-market surveillance (PMS) and vigilance systems, and other regulatory domains impacted by recent changes are the main subjects of the analysis. To assess variations in regulatory strictness, compliance requirements, and approval procedures, comparative synthesis was carried out.
REGULATORY FRAMEWORK:
Indian medical devices regulations: The DCGI of the Central Drugs Standard Control Organization currently regulates scientific instruments as pharmaceuticals. International authorities have faced challenges in the medical device market due to the lack of differentiation between medicine and tools. It is not possible to enter just once. Certain devices are no longer regulated at all, and numerous lists of regulated devices must be combined with laws specific to those devices. India is working to address these issues with a number of new recommendations for clinical entity regulations and a plethora of other, more significant reforms that are still pending within the Indian government.
The CLAA, a division of CDSCO, will attend meetings with medical device advisory committee, which serves as the leading regulatory framework for scientific standardized devices [6]. The CLAA also sets and maintains protective standards, appoints notified bodies, oversees evaluation actions, PMS, issuing of warning letters, and recalls the activities. These policies and reforms promise to clearly unify and expedite the process of producing and importing medical units into India, but they also present their own unique challenges and issues.
The idea for a new regulation had previously been put up for review in 2006. The Medical Devices Regulation Bill of 2006 is the name of the legislation being considered. On December 31, 2009, the new law are going to take effect [7, 8].
Licensing and Registration: The class A and B Devices: State Licensing Authorities (SLA) manage licensing through Notified Bodies' audit assessments.
The class C and D Devices: CDSCO Central Licensing Authorities (CLA) oversees licensing, emphasizing clinical assessment and technical documentation.
Quality Management System (QMS): Manufacturers must comply with ISO 13485 for Quality Management System. This includes:
Design and development control.
Risk management strategies.
Manufacturing process validation.
Premarket Requirements: Device Registration: Required information such as intended use, manufacturing location, and conformity assessment is necessary for market entry.
Clinical Evaluation: Clinical investigations are necessary for Class C and D devices if the available data is inadequate.
Conformity Assessment: Notified Bodies conduct assessments for Class A and B devices, focusing on compliance with ISO standards and MDR guidelines. For Class C and D, CDSCO performs direct evaluations.
Post market Surveillance:
Materiovigilance Program of India (MVPI): Ensures the safety of devices through adverse event monitoring.
Adverse Event Reporting (AER): Mandatory for manufacturers and importers.
Corrective and Preventive Actions (CAPA): Manufacturers must undertake safety actions, including recalls if necessary. [14]
Labeling and UDI: The MDR mandates clear labeling to include:
Unique Device Identification (UDI) for traceability.(14)
Europe: Regulatory Framework by the CE (European Conformity)[9,11]
There was a distinct emphasis on trade and engineering when the first medical device regulations were introduced in Europe. Before the MDD went into effect, a medical device had to meet national standards in each of the several European Member States in order to be sold. As a result, product manufacturers frequently had to deal with several different sets of test requirements [37]. Europe's trade environment became difficult as a result. A type of legislation known as "new approach" legislation was used to lower some of these technical trade barriers. It introduced the idea of a single set of essential requirements that applied to all products and could be supplemented by the use of standards that, when harmonized, allowed a presumption of conformance to those essential requirements.
The use of standards, developed by organizations like the International Standards Organization (ISO), has historically had an engineering focus and has tended to concentrate on processes, testing, and manufacturing methods. Examples of this include the kinds of bench testing necessary to evaluate a family of medical devices.
A conformity assessment is necessary before a medical device can be sold. This can be done directly by the manufacturer of low-risk devices (class I), but it must be done by a notified body for higher-risk devices (class IIa, IIb, and III). Following successful completion, a CE-mark is granted, removing any additional technical trade barriers and enabling marketing in every European Member State.
The MDR has kept this essential system, but it has several novel processes and specific requirements [38]. Early-stage medical device development such as conception, prototyping, and bench or animal testing is unaffected by these changes, but they do bring about a number of significant changes to the clinical research of new medical devices, the evaluation of market entry, and post-market requirements.
Premarket Requirements: Before a product is allowed to be sold, the MDR states that manufacturers need to demonstrate that it is safe and works well.
Technical Documentation: Detailed files that demonstrate that the General Safety and Performance Requirements (GSPR) have been met.
Clinical Evaluation: displaying a substance is safe and helpful in a clinical setting through clinical studies or reviews of the literature
Performance Testing: Verifying that the system works and that the risk reduction measures are in place. [40]
Conformity Assessment: Notified Bodies: Independent organizations chosen by EU member states evaluate the compliance of medium- and high-risk devices.
CE Marking: Devices that satisfy MDR standards are granted the CE Mark, which grants them free market access throughout the EU. (10)
RECENT AMENDMENTS IN MEDICAL DEVICE REGULATIONS:
1. India:
The Central Drugs Standard Control Organization (CDSCO) oversees the Medical Devices Rules (MDR), 2017 and the Drugs and Cosmetics Act, 1940, which govern medical devices in India. The Government of India has implemented a number of regulatory reforms since MDR 2017 was put into effect in order to improve quality assurance, promote domestic manufacturing, strengthen oversight of medical devices, and align Indian regulations with international standards. These changes have concentrated on supply chain monitoring, post-market surveillance, quality management systems, testing infrastructure, and licensing requirements.(11)
Modification of Rules 18 and 19: State Medical Device Testing Laboratories Notification (2023): In 2023, the Ministry of Health and Family Welfare (MoHFW) announced changes to Rules 18 and 19 of the Medical Devices Rules, 2017 to make it easier for State Medical Device Testing Laboratories (MDTLs) to be established and recognized. Medical device testing was mostly dependent on a small number of central laboratories prior to this amendment. The amendment gave state governments the authority to notify labs that could test and assess medical devices in accordance with established guidelines.
Because MDTLs allow for decentralized testing and quicker regulatory action against non-compliant products, the quality assurance framework has been greatly strengthened. These labs assist licensing authorities with post-market surveillance, analytical testing, product specification verification, and complaint investigations. By lessening the load on central testing facilities and guaranteeing prompt detection of faulty or subpar devices, the amendment also enhances regulatory efficiency. (12)
Regulation of Sale and Distribution of Medical Devices (2022): The government made changes in 2022 that mandated that organizations involved in the retail and wholesale sales of medical devices register. The online CDSCO portal was used to implement the registration system, which sought to build an extensive database of distributors, wholesalers, stockists, and retailers involved in the medical device supply chain.
A significant step toward improving accountability and traceability across the distribution network was taken with this amendment. Regulators can keep an eye on the flow of medical devices from producers and importers to healthcare facilities and final consumers by requiring registration. Additionally, product recalls, adverse event investigations, and enforcement actions against unlicensed distributors are made easier by this provision. Additionally, it improves patient safety and public trust in medical technologies by preventing the spread of subpar, unregistered, and counterfeit medical devices.(13)
Exemption for Non-Sterile and Non-Measuring Class A Medical Devices (2022): CDSCO introduced provisions exempting manufacturers of non-sterile and non-measuring Class A medical devices from obtaining manufacturing licenses in recognition of the need to foster innovation and lessen the regulatory burden on low-risk products. Exam gloves, basic surgical instruments, and some assistive devices are examples of class A devices, which are the lowest risk category.
Manufacturers are allowed to self-certify compliance with quality management system requirements under this amendment, but they are still subject to regulatory oversight and inspections. The exemption was implemented as part of the government's "Ease of Doing Business" initiative with the goals of promoting domestic manufacturing, assisting new businesses, and cutting down on administrative delays related to licensing processes. Manufacturers are still in charge of upholding product safety, quality, and compliance with relevant regulations.(14)
Rule 43A (2022) introduction: The addition of Rule 43A, which gives licensing authorities particular authority over license suspension and cancellation, was one of the most important changes to MDR 2017. It was difficult to deal with non-compliance before this amendment because enforcement provisions were less clear.
Law 43A permits licensing authorities to suspend or revoke licenses granted to importers or manufacturers who do not adhere to quality standards, regulatory requirements, or approval conditions. The license holder is typically given a chance to clarify or correct any shortcomings before such action is taken. The amendment guarantees that only compliant products are kept on the market, strengthens regulatory enforcement, and encourages accountability among stakeholders. In the end, Rule 43A improves patient safety by facilitating swift regulatory action against dangerous or subpar medical devices.[15]
Strengthening of Medical Device Testing Infrastructure: India has greatly increased its medical device testing infrastructure in addition to making legislative changes. To help implement MDR 2017, the government has set aside more Central Medical Device Testing Laboratories and Regional Medical Device Testing Laboratories. Additionally, to improve regulatory oversight and inspection operations, Medical Device Officers and qualified technical staff have been appointed.
A thorough assessment of medical devices for safety, performance, biocompatibility, electrical safety, sterility, and other crucial factors is made possible by the growth of testing infrastructure. Additionally, it makes it easier to conduct post-market surveillance, look into unfavorable incidents, and confirm that national and international standards are being followed. Given the growing quantity and complexity of medical devices entering the Indian market, strengthened laboratory capacity is especially crucial. Together, these efforts strengthen the regulatory framework that can guarantee the efficacy, safety, and quality of medical devices over the course of their lifetime. (12)
2. European Union:
Regulations (EU) 2017/745 on Medical Devices (MDR) and (EU) 2017/746 on In Vitro Diagnostic Medical Devices (IVDR) govern medical devices. A more thorough regulatory framework centered on patient safety, clinical evidence, transparency, traceability, and post-market surveillance was established by these regulations, which superseded the earlier Medical Device Directives. To address issues with certification capacity, device shortages, digital registration systems, and emerging technologies like artificial intelligence, the European Union has proposed a number of amendments and implementation measures in recent years.(16)
Regulation (EU) 2024/1860: In response to worries about possible shortages of vital medical devices and delays in conformance assessment processes, Regulation (EU) 2024/1860 was adopted in 2024 to modify both MDR and IVDR. Ensuring the continuous availability of vital medical devices and in vitro diagnostic devices throughout the European Union was one of the amendment's main goals.
In order to give manufacturers more time to comply with MDR and IVDR requirements while preserving market availability, the regulation extended transitional periods for some IVDs and legacy devices. Additionally, it added clauses requiring manufacturers to give advance notice to healthcare facilities and appropriate authorities if they plan to stop or disrupt the supply of essential medical devices. These steps were intended to improve Member States' regulatory readiness and avoid interruptions in patient care.(17)
Progressive Roll-Out of EUDAMED: One of the most important regulatory innovations implemented under MDR and IVDR is the European Database on Medical Devices (EUDAMED), which operates as a centralized digital platform that integrates data about manufacturers, authorized representatives, importers, notified bodies, certificates, clinical investigations, vigilance operations, market surveillance, and device registration.
In order to facilitate stakeholder adoption, the database is being implemented in stages. By making crucial information about medical devices sold in the EU accessible to regulators, medical professionals, and the general public, EUDAMED enhances transparency. Additionally, it facilitates coordinated regulatory oversight among Member States and enhances traceability through the integration of Unique Device Identification (UDI) systems. (18)
Supply Shortage Reporting Requirements: Regulation (EU) 2024/1860, a significant amendment, mandates the reporting of anticipated supply disruptions. Before stopping or interrupting the supply of goods deemed necessary for patient care, manufacturers of critical medical devices and in vitro diagnostic devices must notify the appropriate authorities.
Depending on the device's characteristics and the situation, the notification period typically lasts between six months and a year. Authorities are able to evaluate the possible effects on healthcare systems, plan mitigation measures, find substitute suppliers, and lower the possibility of patient shortages thanks to this requirement. The amendment takes into account the COVID-19 pandemic's lessons about supply chain vulnerabilities and healthcare readiness.[17]
Support for Orphan Medical Devices (2024): The European Medicines Agency (EMA) started a pilot program in 2024 with the goal of providing support for orphan medical devices meant for uncommon illnesses and ailments that only affect a small number of patients. Due to their small market size, lack of clinical data, and diminished commercial incentives, these devices frequently face significant development challenges.
Through expert panels, the program offers manufacturers and notified bodies regulatory guidance and scientific advice throughout the development and conformance assessment process. While promoting research and development in areas of unmet medical need, the initiative aims to increase patients with rare diseases' access to cutting-edge technologies.[19]
Connectivity to the European Union Artificial Intelligence Act: With the passage of Regulation (EU) 2024/1689, also referred to as the Artificial Intelligence Act, medical devices with AI capabilities now face additional regulatory requirements. High-risk AI systems include many software-based medical devices that use diagnostic AI applications, clinical decision support systems, predictive analytics, and machine learning algorithms.
Additional regulations pertaining to risk management, data quality, transparency, cybersecurity, human oversight, algorithm validation, and post-market monitoring must be followed by manufacturers of such devices. The AI Act's integration with MDR establishes a dual regulatory framework that guarantees the safety of medical devices as well as the ethical application of AI in healthcare.[20]
Mandatory Implementation of EUDAMED Modules (2025–2026 Amendment): Following the publication of Commission Decision (EU) 2025/2371 in November 2025, one of the biggest recent developments took place. In accordance with Regulation (EU) 2024/1860's transitional provisions, the European Commission formally announced that the first four EUDAMED modules had reached full functionality and fulfilled all necessary technical requirements. (21)
As a result, all relevant economic operators, including manufacturers, authorized representatives, importers, and notified bodies, were required to use the four main EUDAMED modules starting on May 28, 2026. The following modules are required:
• Actor Registration
• UDI/Device Registration
• Notified Bodies and Certificates
• Market Surveillance
This amendment represents a major milestone in the digital transformation of the European medical device regulatory system. Mandatory use of EUDAMED strengthens transparency, improves traceability of medical devices, enhances information exchange among regulatory authorities, and facilitates more efficient post-market surveillance activities. It also requires manufacturers to maintain accurate electronic records and comply with expanded reporting obligations throughout the product lifecycle. (22)
Strengthening of UDI and Device Registration Requirements (2026): Following mandatory activation of EUDAMED modules in 2026, manufacturers are required to register device information using the Unique Device Identification (UDI) system. The UDI framework enables each medical device to be assigned a unique identifier, improving product traceability, recall management, vigilance reporting, and supply chain monitoring. Manufacturers must submit UDI and device-related information through the EUDAMED UDI/Device Registration Module before placing products on the EU market. The registration requirements also apply to legacy devices covered under transitional provisions. This amendment significantly strengthens lifecycle monitoring and supports rapid identification of affected devices during safety investigations or field corrective actions.(23)
Emerging Regulatory Support for Breakthrough Medical Devices (2026): In 2026, EMA introduced an additional pilot programme providing expert panel support for breakthrough medical devices (BtX). The initiative offers scientific and regulatory guidance for manufacturers developing highly innovative devices with significant clinical benefits. The programme supports manufacturers during clinical strategy development and conformity assessment planning while facilitating communication between manufacturers and notified bodies. The objective is to accelerate patient access to innovative technologies while maintaining compliance with MDR requirements and ensuring robust evidence of safety and performance.(24)
Phased licensing implementation (2020–2023): India established a risk-based phased licensing framework to facilitate industry adaptation.
The stage of Registration:
Licensing Conditions for Various Device Classes: Risk classification guided the implementation of licensing requirements:
1. Class A and B (Moderate to Low Risk):
2. Moderate to High Risk Classes C and D:
Timeline of Implementation:
This methodical approach enhanced risk-based oversight, regulatory effectiveness, and resource allocation.
Enhancing Quality and Testing Infrastructure: The most recent modifications place a strong emphasis on enhancing systems for conformity assessment and quality assurance.
The designation and reinforcement of Medical Device Testing Laboratories (MDTLs) for standardized testing was started by CDSCO [27].
MDTLs carry out:
These labs improve the ability to make and enforce regulations [29].
Qualifications for Quality Certification: ISO 13485:2016 must be implemented for Quality Management Systems (QMS) [30]. adherence to the Fifth Schedule of MDR 2017 regarding Good Manufacturing Practices (GMP). CDSCO and SLAs conduct routine audits and inspections [31
A greater focus on:
These actions enhance product quality and bring Indian manufacturers into compliance with international regulatory standards [32].
Digital Regulatory Reforms: A key element of the most recent regulatory changes has been digital transformation.
Online Licensing System:(33)
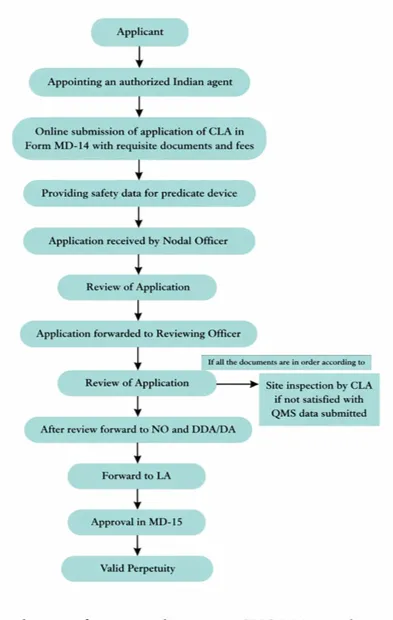
Fig 1: Application for import license via SUGAM portal
Benefits include:
National Single Window System (NSWS): Integration of medical device approvals into the National Single Window System (NSWS) [35].
Provides a centralized platform for regulatory clearances, including:
Enhances:
Medical device approval pathway in India(39)

Fig 2: Medical device approval pathway in India
Medical device approval pathway in Europe[3,44]
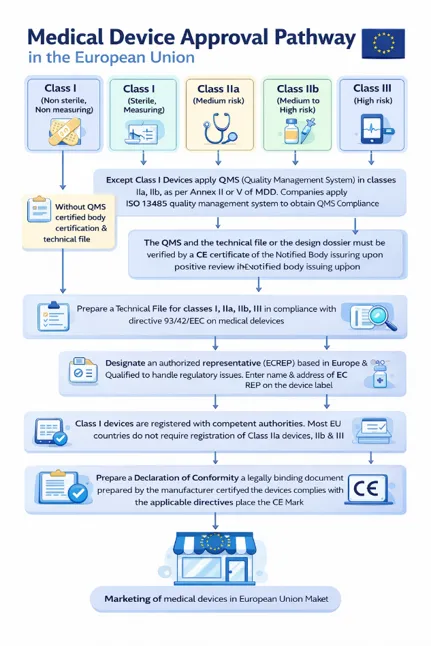
Fig 3: Medical device approval pathway in Europe
COMPARATIVE STUDIES (40)
Table No 5: – Comparative Review on Medical Device Regulation Based on Geography
|
Sr. No. |
Comparatives |
India |
Europe |
|
1 |
Regulatory authority |
DCGI under CDSCO |
EMA & RA of Member State |
|
2 |
Regulation |
Medical Device Rules, 2017 |
Regulation (EU) 2017/745 |
|
3 |
Class of Medical device |
Four classes: Class A, Class B, Class C, Class D |
Three classes: Class I, Class IIa, Class IIb, Class III |
|
4 |
Regulatory Pathway |
Market Authorization application to competent authority |
Multiple pathways for different classes |
|
5 |
Type of application |
Marketing Authorization Application |
Marketing Authorization Application |
|
6 |
Guidelines / Regulations |
Schedule M, Schedule M III – GMP, Schedule Y – Clinical Trials |
Directive 93/42/EEC – Medical Device Directive, 93/79/EC – CE Marking |
|
7 |
Application Submission |
Electronic Submission |
eSubmission Gateway / Web Client |
|
8 |
QMS Requirement |
ISO 13485:2016 |
ISO 13485 or as needed under 94/42/EEC |
|
9 |
Assessment of technical data |
Notified Bodies (NB) under CDSCO |
Notified Bodies (NB) under National Regulatory Authority |
|
10 |
Establishment Registration Requirement |
Premises Registration |
Premises Registration |
|
11 |
Data Presentation (Electronic/Paper) |
Paper / Electronic |
Electronic / Paper submission |
|
12 |
Language |
English |
English |
|
13 |
Clinical test reports |
Mandatory for Class C and D devices |
Required for Class IIb and III medical devices |
|
14 |
Fees for Pathway |
Manufacturing License fee |
Fees for available pathways |
|
15 |
Registration expiry |
5 years from date of approval. |
Lifetime for Class I. CE mark validity is 3 years for Class IIa, IIb, and III. |
|
16 |
Time required for approval (in Months) |
6–12 months |
Class I (1 month); Class II (3–6 months); Class III (9–15 months). |
|
17 |
Final Outcome |
Manufacturing License |
CE Mark with NB number |
|
18 |
On-site audits |
Applicable |
Applicable |
Recommendations for Regulatory Improvements in India(41)
|
Focus Area |
Recommended Actions |
|
1. Post-Market Surveillance |
Make periodic reviews mandatory • Integrate AI-based early warning systems |
|
2. Clinical Trials |
Increase local clinical studies • Reduce reliance on foreign data |
|
3. Transparency & Efficiency |
Standardize approval timelines • Create fast-track pathways for Class C & D devices |
|
4.Efficiency |
Class C and D devices |
FUTURE DIRECTIONS:
The regulations governing the medical device industry must evolve along with it in order to ensure that it is safe, effective, and able to reach global markets. India has the chance to improve its regulatory procedures through policy improvements, international cooperation, and simplified market entry tactics, given its expanding medical technology industry.
I. Strengthening Policy Enhancements: A combination of the Medical Devices Rules, 2017, India's medical device laws have advanced significantly; however, more improvements could improve effectiveness and openness:
II. Global Harmonization for Better Compliance: India can benefit from closer alignment with international regulatory standards, reducing redundancies and facilitating faster approvals.
III. Improving Market Entry Strategies: India needs to become a world leader in medical devices. innovation, regulatory pathways must be neutralized to good investment, research, and international trade.
India's medical device regulations are currently at a turning point. India can lower obstacles, improve patient safety, and strengthen its ties to the rest of the world through policy reform, global harmonization, and industry-friendly initiatives. Enhancing clinical evaluation, post-marketing surveillance, and industrial cooperation stand out as potential motivators to maintain India's competitiveness in promoting medical innovation and access.
CONCLUSION
Regulations pertaining to medical devices are essential for guaranteeing the efficacy, performance, safety, and quality of medical technologies. This comparative analysis shows that in order to improve patient safety, bolster oversight, and harmonize their regulatory frameworks with changing international norms, both the European Union and India have recently implemented substantial regulatory reforms..
After the Medical Devices Rules, 2017 were put into effect and later amended, India made significant strides. The country's regulatory framework has been greatly enhanced by important reforms, such as the expansion of notified medical devices, mandatory licensing requirements, strengthening of testing infrastructure, establishment of Medical Device Testing Laboratories (MDTLs), improvement of post-market surveillance mechanisms, and digitalization of regulatory processes through the National Single Window System and SUGAM portal. In addition to promoting domestic manufacturing under the "Make in India" initiative, these measures have improved accountability, transparency, quality assurance, and ease of doing business.
On the other hand, through MDR 2017/745 and IVDR 2017/746, the European Union has created a more developed and comprehensive regulatory framework. This framework has been reinforced by recent amendments like Regulation (EU) 2024/1860, the implementation of EUDAMED, increased UDI requirements, supply shortage reporting obligations, and integration with the Artificial Intelligence Act. These changes place a strong emphasis on proactive risk management, traceability, transparency, lifecycle monitoring, and the creation of clinical evidence. Particularly in the areas of clinical evaluation, post-market surveillance, and digital regulatory infrastructure, the EU system still sets the standard for regulatory rigor worldwide.
The comparative analysis shows that although both jurisdictions prioritize patient safety and take a risk-based approach, there are still significant differences in terms of clinical evidence requirements, approval timelines, documentation standards, regulatory complexity, and conformity assessment processes. While India's regulatory system is quickly moving toward international harmonization through the adoption of ISO 13485, IMDRF principles, and enhanced regulatory oversight, the EU framework is still more strict and comprehensive.
All things considered, the recent changes in both areas share the goal of guaranteeing high-quality, safe, and efficient medical devices while encouraging innovation and market accessibility. Future challenges posed by emerging technologies like artificial intelligence, software as a medical device (SaMD), and personalized healthcare solutions will require ongoing investment in regulatory infrastructure, digital transformation, international harmonization, and strong post-market surveillance. Both India and the EU are anticipated to make major contributions to the creation of a globally harmonized medical device regulatory environment that fosters innovation while upholding the highest standards of patient protection as regulatory convergence grows.
REFERENCES


Pratima Shinde, N. B Chalmale, Dr. Satpute K. L, Chaitanya Shinde, Anjali Dudhate, Sakshi Swami, Comparative Evaluation of Recent Regulatory Amendments for Medical Devices in India and EU, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 6247-6264. https://doi.org/10.5281/zenodo.20839350
 10.5281/zenodo.20839350
10.5281/zenodo.20839350