
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Guru Nanak College of Pharmaceutical Sciences, Dehradun
Drug recalls are a critical public health intervention designed to remove defective, adulterated, or misbranded pharmaceutical products from the market. As pharmaceutical supply chains become increasingly globalised and the volume of recalls rises in both developed and emerging economies, understanding the strengths and weaknesses of different regulatory frameworks is essential for improving global drug safety. This comparative study examines the drug recall procedures of the United States and India—two nations that play interconnected yet contrasting roles in the international pharmaceutical landscape. The USA, with its centralized regulatory system under the Food and Drug Administration (FDA), offers a mature, legally enforceable recall framework codified in 21 CFR Part 7, supported by robust seizure, injunction, and inspection powers. India, through the Central Drugs Standard Control Organization (CDSCO), operates under a federal structure with shared authority between the centre and state licensing authorities. Despite having issued recall guidelines in 2012 (revised 2017) that mirror the three tier classification of the FDA, these guidelines remain non binding administrative instructions lacking statutory force. The study analyses governance structures, statutory authorities, key legal provisions, enforcement mechanisms, and the unique challenges posed by India’s multi state environment. Findings reveal that while the FDA’s system benefits from centralised authority, direct enforcement powers, and statutory transparency requirements, the CDSCO’s recall regime is characterised by fragmented state level implementation, limited legal teeth, and heavy reliance on voluntary manufacturer cooperation. The absence of a statutory recall provision in the Drugs and Cosmetics Act, combined with non incorporated guidelines, undermines the effectiveness and reliability of recalls in India. The study concludes that meaningful reform in India requires embedding recall mandates into primary legislation, establishing a centralized public recall database, and strengthening centre state coordination mechanisms. For the USA, ongoing challenges include enforcement against foreign manufacturers and managing drug shortages following large scale recalls. The comparative analysis offers transferable lessons for regulatory convergence under the International Council for Harmonisation and the World Health Organization.
The critical role of drug recalls in protecting public health from defective or harmful pharmaceutical products cannot be overstated. In an era where pharmaceutical supply chains span continents and millions of patients depend on timely access to safe medicines, the ability to swiftly and effectively remove a compromised product from the market is a cornerstone of modern drug safety regulation[1]. A drug recall is not merely a logistical exercise; it is a public health intervention designed to prevent morbidity and mortality by eliminating products that fail to meet quality, safety, or efficacy standards. These failures can arise from diverse causes—manufacturing errors, contamination, incorrect labelling, packaging defects, or unexpected adverse reactions identified only after marketing approval. Without a robust recall mechanism, such defects can propagate through the distribution network, reaching hospitals, pharmacies, and ultimately patients, with potentially catastrophic consequences. The 2008 heparin contamination incident in the United States, which led to dozens of deaths and hundreds of serious allergic reactions, exemplifies the lethal potential of a delayed or ineffective recall. Conversely, a well-executed recall—such as the rapid withdrawal of valsartan products contaminated with probable human carcinogens (NDMA) in 2018—demonstrates how regulatory vigilance can mitigate harm, even when the initial quality failure originates in a distant manufacturing facility. Thus, drug recall procedures are not a peripheral aspect of pharmaceutical regulation; they are a vital safety net that catches defects that have escaped earlier checks in the drug development and manufacturing chain. The increasing volume and complexity of drug recalls in both developed and emerging economies further underscore the urgency of understanding and improving these procedures[2]. Over the past two decades, the number of drug recalls reported annually to major regulators has risen significantly, a trend driven by several interrelated factors. First, the globalization of pharmaceutical production has created long, opaque supply chains. Active pharmaceutical ingredients (APIs) and finished dosage forms are often manufactured in one country, packaged in another, and distributed globally. This fragmentation introduces multiple points of failure, from cross-contamination in shared facilities to the use of adulterated raw materials. Second, advances in analytical chemistry have lowered the detection limits for impurities, enabling regulators and manufacturers to identify previously undetectable or ignored contaminants—such as nitrosamines—triggering widespread recalls that would have been impossible to initiate a generation ago. Third, heightened pharmacovigilance and adverse event reporting systems, coupled with social media and 24‑hour news cycles, increase the pressure on firms to act quickly when a potential defect is identified. In the United States, the Food and Drug Administration (FDA) processed over 1,200 drug recall events in 2023 alone, ranging from minor labelling corrections to Class I recalls involving life‑threatening risks[3]. India, as a major global supplier of generic medicines, has also seen a steep rise in recall activity, both voluntarily initiated by domestic manufacturers and mandated by importing countries. However, the complexity of recalls is not only quantitative; it is also qualitative. Recalls now frequently involve not just single products but entire families of drugs, requiring coordination across multiple regulatory jurisdictions, languages, and legal systems. Moreover, the rise of biologics and biosimilars introduces new challenges, as these products are more sensitive to storage and handling deviations, and their recall often forces patients to switch therapies abruptly—a decision with its own clinical risks. Hence, studying the procedural frameworks that govern recalls is not an academic luxury; it is a practical necessity for ensuring that the volume and complexity of modern drug defects do not outpace the capacity of regulatory systems to respond[4][5].
Why a comparative analysis of India and the USA is relevant for improving global drug safety can be understood by examining the distinctive yet interconnected roles these two nations play in the international pharmaceutical landscape. The United States represents the world’s largest pharmaceutical market by revenue, with a regulatory system that is often considered a gold standard for drug safety. The FDA’s recall procedures, embedded in 21 CFR Part 7 and elaborated in the Regulatory Procedures Manual, benefit from decades of refinement, significant legal authority, substantial budgetary resources, and a culture of transparency through publicly accessible enforcement reports. The US system is predominantly voluntary—firms initiate most recalls—but the FDA can leverage its inspection, seizure, and injunction powers to compel action when necessary. For any other country seeking to design or upgrade its recall regulations, the US model provides a rich source of best practices, as well as lessons from its own failures and limitations, such as the difficulty of enforcing recalls on foreign facilities or the risk of drug shortages following large‑scale withdrawals[6][7].
In contrast, India is the “pharmacy of the world,” supplying over 60% of global vaccine demand and 20% of generic medicines by volume, including a substantial fraction of drugs consumed in the United States. Yet India’s domestic recall framework has long been criticized as fragmented and legally weak. The Central Drugs Standard Control Organization (CDSCO) operates under a federal structure where state licensing authorities retain significant autonomy, and the official “Guidelines on Recall and Rapid Alert System” have not been incorporated into the Drugs and Cosmetics Rules, rendering them non‑binding. Consequently, while India has produced recall guidelines that mirror the three‑tier classification (Class I, II, III) used by the FDA, implementation remains inconsistent. Recalls are largely voluntary, and there is no centralized, publicly searchable database of all drug recalls occurring within India[8][9]. This creates a troubling asymmetry: drugs that fail quality tests in India may not be systematically recalled from the Indian market, yet the same drugs destined for export are subject to the stringent recall requirements of importing countries like the US or the European Union. A comparative analysis thus illuminates a fundamental tension in global drug safety governance: supply chains are global, but recall authority remains national or sub‑national. By juxtaposing a mature, legally enforceable system (USA) with a rapidly evolving but structurally constrained system (India), researchers can identify which elements of recall procedure are truly essential and transferable across different legal and economic contexts. For India, the comparison offers concrete pathways for legislative reform—such as embedding recall mandates into the Drugs and Cosmetics Rules and establishing a public recall registry. For the United States, the comparison highlights the risks of relying on foreign manufacturers that operate under weaker domestic recall regimes, reinforcing the need for mutual recognition agreements and joint inspection programs. Ultimately, improving drug safety on a global scale requires not only strong individual regulators but also harmonized expectations and cooperative mechanisms. A detailed comparative study of India and the USA serves as a microcosm of this larger challenge, offering lessons that can inform regulatory convergence under the auspices of the International Council for Harmonisation (ICH) and the World Health Organization (WHO)[10][11].
Historical Evolution of Drug Regulation
The historical evolution of drug regulation in India, particularly the seminal event leading to the Drugs and Cosmetics Act of 1940 and Rules of 1945, is often traced to the Maselkar poisonings of 1937—though the name is often misremembered in popular accounts. In fact, the tragedy that galvanized Indian drug law was the 1937 poisoning in the United States from Elixir Sulfanilamide, which killed over 100 people, and its ripple effects on British India[12][13]. However, the most directly cited Indian catalyst is the 1937 death of 14 patients at the Masulipatnam Government Hospital (present‑day Machilipatnam, Andhra Pradesh) after being administered a contaminated analgesic and antipyretic mixture. Some historical records refer to this as the “Maselkar” or “Masulipatnam” poisoning. The incident occurred when a batch of a proprietary medicine called “M&B 693” (a sulfonamide derivative) was improperly manufactured, leading to severe adverse reactions. Although the exact number of victims remains debated, the public outrage forced the colonial government to recognize the utter inadequacy of the then‑prevailing law—the Poisons Act of 1919, which focused narrowly on a limited set of labeled poisons and did not regulate the quality, safety, or efficacy of therapeutic drugs. In response, the government appointed a committee chaired by Lieutenant Colonel R. N. Chopra, a renowned pharmacologist, to draft comprehensive drug legislation. The Chopra Committee’s report, submitted in 1939, became the blueprint for the Drugs and Cosmetics Act, 194[14][15]0. The Act was passed by the Central Legislative Assembly and received assent on 10 April 1940, but its operative rules—the Drugs and Cosmetics Rules—were not finalized until 1945, after a series of further consultations and the disruption caused by World War II. The Act established the concept of “standard quality” for drugs, created schedules for different categories of medicines, and mandated that manufacturers obtain licenses. Importantly, it also empowered the government to prohibit the manufacture and sale of any drug that did not conform to specified standards. This foundational legislation, although amended dozens of times over the ensuing eight decades, remains the statutory backbone for drug regulation in India. The 1940 Act did not, however, explicitly provide for a structured drug recall procedure as understood today; recalls were handled on an ad hoc basis through licensing actions and court injunctions. It was not until the 1970s and 1980s, following international developments and high‑profile contaminations (e.g., the 1982 Tylenol cyanide incident in the US), that Indian authorities began formulating voluntary recall guidelines. The current “Guidelines on Recall and Rapid Alert System” were first issued in 2012 and revised in 2017, but they still lack the force of law—a deficiency that directly motivates contemporary comparative research[16]h.
For the United States, the historical evolution of drug regulation is similarly punctuated by a catastrophic mass poisoning that reshaped federal authority[17]: the Elixir Sulfanilamide disaster of 1937. In the autumn of that year, the S. E. Massengill Company, a Tennessee‑based manufacturer, prepared a liquid formulation of the new sulfanilamide antibiotic using diethylene glycol (DEG) as a solvent. Diethylene glycol, a cheap and effective solvent, is also a potent toxin that causes acute renal failure and metabolic acidosis[18][19]. The company had conducted no safety testing of the solvent; indeed, at the time, there was no federal requirement for pre‑marketing safety evaluation of drugs. The resulting “Elixir Sulfanilamide” was distributed across the United States. Within weeks, physicians began reporting patients—many of them children—who developed agonizing abdominal pain, anuria, and coma followed by death. The final death toll reached 107, including many young patients who had taken the elixir for minor streptococcal infections. This disaster triggered a national outcry and directly propelled the passage of the Federal Food, Drug, and Cosmetic (FD&C) Act of 1938. Unlike the earlier Pure Food and Drug Act of 1906, which focused primarily on prohibiting misbranding and adulteration after the fact, the 1938 Act introduced several landmark provisions: it required that drugs be proven safe before marketing (though not yet effective); it authorized factory inspections; it extended control to cosmetics and therapeutic devices; and it provided for court injunctions and seizure of illegal products[20][21]. The 1938 Act did not, however, establish a mandatory recall procedure. Recalls remained essentially voluntary, with the FDA’s main leverage being the threat of seizure or injunction. The absence of a formal recall power became increasingly problematic as the pharmaceutical industry expanded after World War II. A second transformative event—the thalidomide tragedy of the early 1960s, in which the drug caused thousands of birth defects in Europe and elsewhere—exposed the safety gaps even in the 1938 framework. Although thalidomide was never approved for general use in the United States, largely due to the vigilant review of FDA medical officer Frances Oldham Kelsey, the near‑miss galvanized Congress to pass the Kefauver‑Harris Amendments in 1962. These amendments radically reshaped drug regulation by requiring not only safety but also “substantial evidence” of efficacy from adequate and well‑controlled clinical trials. They also mandated that pharmaceutical companies report adverse events to the FDA, gave the agency explicit authority to withdraw approvals, and required informed consent from research subjects. In terms of recalls, the 1962 amendments strengthened the FDA’s hand by allowing the agency to monitor and oversee voluntary recalls more closely and, for the first time, to request recall of defective prescription drugs under the agency’s expanded injunction powers[22]. Yet it was not until 1974, after a series of voluntary recalls proved ineffective in removing contaminated intrauterine devices and other products from the market, that the FDA formally issued its “Recalls (Including Product Corrections) – Policy, Procedures, and Industry Responsibilities” (21 CFR Part 7). This regulation codified the now‑familiar three‑class recall system (Class I, II, III) based on relative health hazard and detailed the respective responsibilities of the FDA and the recalling firm. Unlike India, where recall guidelines remain non‑binding, the US framework is enforceable law, with civil and criminal penalties for non‑compliance[23][24].
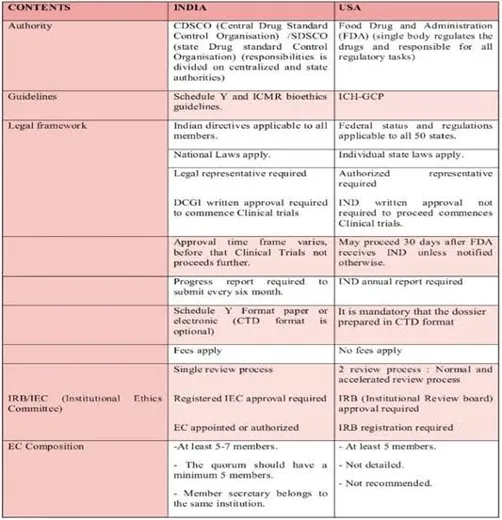
Fig: 1 US vs. India Regulatory Comparison
The historical evolution thus reveals a striking parallel: in both countries, tragedy triggered legal reform; but while the United States continued to build enforceable recall authority through successive legislative amendments, India’s progress has stalled at the stage of non‑statutory guidelines[25][26]. This divergence is central to any comparative study, as it explains why recalls in India remain less transparent, less systematic, and less reliable than those in the US—despite India’s outsized role in global drug supply. Recognizing this historical context is essential for understanding not only the present limitations of the Indian system but also the feasible pathways for its future improvement, drawing on the hard‑won lessons of the US experience.
The U.S. Food and Drug Administration (FDA)
The U.S. Food and Drug Administration (FDA) operates as a centralized, unified federal agency with comprehensive authority over drug safety, a structural feature that fundamentally shapes its recall procedures[27][28]. Several key characteristics define the FDA’s role in drug recalls:
Centralized governance structure – Unlike federal systems where power is diffused among states, the FDA derives its authority directly from the federal government and exercises jurisdiction over all 50 states, the District of Columbia, and U.S. territories. This centralization means that a recall decision made by the FDA applies uniformly across the entire nation, eliminating the possibility of a drug being recalled in one state but remaining available in another. The agency’s field offices, organized into five geographic regions, implement recall actions under a single command structure, ensuring consistency in classification, communication, and enforcement[29][30].
Legal basis in the FD&C Act – The Federal Food, Drug, and Cosmetic Act (FD&C Act) of 1938, as amended, serves as the statutory foundation for all FDA drug safety activities. The Act grants the FDA explicit authority to regulate drugs from pre‑market approval through post‑market surveillance. Relevant to recalls, Section 501(a)(2)(B) defines a drug as “adulterated” if it has been manufactured, processed, packed, or held in conditions that do not conform to Current Good Manufacturing Practice (CGMP). Section 502 defines misbranding violations. Once a drug is deemed adulterated or misbranded, the FDA can pursue seizure (Section 304), injunction (Section 302), or criminal prosecution (Section 303). These legal remedies provide powerful leverage to encourage voluntary recalls[31].
Codified recall procedures in 21 CFR Part 7 – The specific rules governing recalls are codified in Title 21 of the Code of Federal Regulations, Part 7 (21 CFR Part 7), subpart C (“Recalls (Including Product Corrections) – Policy, Procedures, and Industry Responsibilities”). This regulation defines key terms such as “recall,” “market withdrawal,” and “medical device safety alert.” It establishes the three‑class system (Class I, II, III) based on the relative health hazard. Critically, 21 CFR Part 7 formalizes the FDA’s role in monitoring and evaluating recalls, including the requirement for firms to submit recall strategies, periodic progress reports, and recall effectiveness checks. Although recalls are typically voluntary in initiation, the regulation empowers the FDA to request a recall and, if the firm refuses, to seek a court order mandating the recal[32][33]l.
Statutory recall authority vs. voluntary practice – It is a common misconception that the FDA cannot mandate recalls. Under Section 518(e) of the FD&C Act (for medical devices) and Section 423(f) (for foods, including dietary supplements), the FDA has limited mandatory recall authority. For drugs, however, the FDA does not have a standalone mandatory recall power; instead, it relies on its authority to seize products and enjoin manufacturing. In practice, firms almost always comply with an FDA request for a voluntary recall because refusal invites seizure, injunction, or criminal penalties. Thus, the system is “voluntary” in name but effectively compulsory due to the FDA’s robust enforcement toolkit[34].
Global reach through import alerts – The FDA’s authority extends beyond U.S. borders. Under Section 801(a) of the FD&C Act, the FDA can detain imported drugs that appear to be adulterated or misbranded. For foreign facilities that repeatedly violate CGMP or fail to cooperate in recalls, the FDA can issue an import alert, which allows detention without physical examination of all products from that facility. This extraterritorial reach is particularly important given that most drug recalls originate from foreign manufacturers, including many in India[35].
In summary, the FDA’s recall system benefits from centralized governance, a strong statutory framework, codified procedures, effective leverage for voluntary compliance, and international enforcement tools. These features make the U.S. system a benchmark for other regulators, despite ongoing challenges related to global supply chains[36].
India’s Central Drugs Standard Control Organization (CDSCO)
India’s Central Drugs Standard Control Organization (CDSCO) serves as the apex regulatory authority for drugs and cosmetics, but it operates within a federal structure that significantly constrains its ability to implement uniform recall procedures. The following points describe the CDSCO’s governance framework and the unique challenges it faces:
Apex but not exclusive authority – The CDSCO, headed by the Drugs Controller General of India (DCGI), is the central regulatory body responsible for approving new drugs, conducting clinical trials, setting standards for drugs, and coordinating with state regulators. However, the Drugs and Cosmetics Act, 1940 and its Rules, 1945 create a shared responsibility between the central and state governments. Under the Act, state governments appoint their own State Licensing Authorities (SLAs), who are responsible for issuing manufacturing licenses, inspecting facilities, and taking enforcement actions – including recall‑related actions – within their respective states.
Legal basis: Drugs and Cosmetics Act, 1940 & Rules, 1945 – The Act and Rules constitute the primary legal framework. The Act defines “misbranded drugs,” “adulterated drugs,” “spurious drugs,” and “not of standard quality” (NSQ) drugs. Section 26A of the Act, inserted in 2005, empowers the central government to prohibit the manufacture, sale, or distribution of any drug in the public interest. This section is often cited as the statutory basis for mandatory recalls, as it allows the CDSCO to order a ban on a specific drug. However, Section 26A has been used sparingly, and the process is cumbersome, requiring the central government to issue a notification in the Official Gazette. Unlike the FDA’s seizure powers, Section 26A does not provide a mechanism for physically removing already distributed products from the market – it only prohibits further sale[37].
Non‑binding recall guidelines – In 2012, the CDSCO issued the “Guidelines on Recall and Rapid Alert System,” revised in 2017. These guidelines adopt a three‑class system (Class I, II, III) similar to the FDA’s and outline responsibilities for manufacturers, importers, wholesale and retail pharmacists. They require firms to designate a recall coordinator, maintain distribution records, conduct mock recalls annually, and notify the CDSCO within 24 hours for a Class I recall. However – and this is a critical limitation – these guidelines have never been incorporated into the Drugs and Cosmetics Rules through a formal amendment. Consequently, they are non‑binding administrative instructions, not law. A manufacturer who refuses to initiate a voluntary recall cannot be prosecuted solely for violating the guidelines, although the underlying NSQ drug may still be actionable under other sections of the Act[38].
The unique challenge of implementing recalls in a multi‑state environment – The federal distribution of powers creates several practical difficulties. First, manufacturing licenses are issued by state authorities, meaning that a drug recalled by the CDSCO may still be legally manufactured under a state license unless that state SLA cooperates. Second, distribution networks cross state boundaries; a recall strategy that works in Maharashtra may fail in West Bengal if the local SLA does not enforce it.[39]
Fig: 2 Regulatory Affairs and Intellectual Property Rights in Immunotherapeutics
Voluntary recall as the dominant practice – In the absence of a binding recall statute, most recalls in India are initiated voluntarily by responsible manufacturers, often in response to a non‑conformity detected through their own quality systems or after a warning from an importing country (e.g., a USFDA import alert). The CDSCO can publicly announce a recall on its website, but it cannot compel pharmacies or hospitals to return the product without state-level enforcement. This voluntary system works reasonably well for large, reputable Indian pharmaceutical companies that export to regulated markets, but it provides little protection against smaller, less scrupulous manufacturers who may simply ignore recall requests.
In essence, the CDSCO’s recall system is characterized by an apex coordinating body with limited legal teeth, a federal structure that fragments enforcement authority, non‑statutory guidelines, and heavy reliance on voluntary cooperation from both industry and state regulators. This configuration explains why, despite having recall guidelines since 2012,[40] India still lacks a systematic, transparent, and enforceable recall mechanism comparable to that of the United States.
Comparative Governance Analysis
A direct comparison of governance structures, statutory authorities, and key statutes between India and the United States reveals fundamental differences that explain the divergent effectiveness of drug recall procedures in the two countries. The following points synthesize the comparative analysis:
A. Structure: Centralized (USA) vs. Federal (India)
United States (centralized): The FDA operates as a single, unified federal agency with nationwide jurisdiction. There is no layer of state drug licensing authorities that can override or ignore federal recall decisions. Field offices are administrative sub‑units of the FDA, not independent state regulators. This centralization ensures that once the FDA classifies a recall, the decision is binding across all states, territories, and distribution channels. Uniformity is further reinforced by federal pre‑emption – state laws that conflict with federal drug safety requirements are generally invalid. Consequently, a drug recalled in California is automatically recalled in Texas and New York.
India (federal): The CDSCO is the central authority, but the Constitution of India places “drugs and poisons” in the Concurrent List (List III, Entry 19), meaning both Parliament and state legislatures can legislate on the subject. The Drugs and Cosmetics Act is a central law, but its implementation relies heavily on state licensing authorities, which are independent bureaucracies accountable to their respective state governments. There is no automatic pre‑emption; states can, in theory, adopt stricter or more lenient enforcement practices as long as they do not directly contradict the central law. This federal asymmetry means that a recall direction from the DCGI may be implemented promptly in a state with a well‑resourced drug control department, but delayed or ignored in a state with limited capacity or competing political priorities. The result is a patchwork recall landscape that undermines patient safety[41].
B. Authority: FDA’s statutory powers vs. CDSCO’s reliance on state cooperation
United States (direct statutory powers): The FDA possesses direct, enforceable legal authority under the FD&C Act. It can inspect factories without prior notice (Section 704), detain suspect products administratively (Section 304(g)), seize adulterated or misbranded drugs (Section 304(a)), and seek injunctions to stop manufacturing or distribution (Section 302). Although the FDA cannot issue a unilateral mandatory recall order for drugs, its seizure and injunction powers create such strong leverage that firms almost always comply with voluntary recall requests. Additionally, the FDA can impose civil monetary penalties, initiate criminal prosecutions (Section 303), and issue debarment orders against corporate officers. These powers are exercised directly by federal employees, not delegated to state officials.
India (indirect, cooperative authority): The CDSCO lacks comparable direct enforcement tools. While Section 26A of the Drugs and Cosmetics Act allows the central government to prohibit a drug, this is a rarely used, cumbersome administrative process that does not include provisions for physically retrieving distributed stock. The primary enforcement actions – suspension or cancellation of licenses, seizures, and prosecutions – are carried out by State Licensing Authorities under state rules. The CDSCO can request, coordinate, and publish recall notices, but it cannot compel a state SLA to act. If a state chooses not to enforce a recall, the CDSCO’s only recourse is to refer the matter to the central government for possible direction under Article 256 of the Constitution (which is rarely invoked in drug safety matters). Thus, the CDSCO’s authority is fundamentally cooperative and advisory, not coercive.
C. Key Statutes: Comparison of the FD&C Act and the Drugs and Cosmetics Act
The FD&C Act is a comprehensive, living statute that has been amended dozens of times to address emerging challenges, including the Kefauver‑Harris Amendments (1962) for efficacy, the Prescription Drug Marketing Act (1987) for wholesale distribution, and the Drug Supply Chain Security Act (2013) for traceability. The Drugs and Cosmetics Act, while regularly amended, has not undergone similarly foundational overhauls in recall‑related provisions. Key definitions (e.g., “not of standard quality”) are broad but lack specific recall triggers[42].
The FD&C Act does not have a dedicated “recall section” for drugs, but it provides a suite of powers (seizure, injunction, detention, import alerts) that functionally enable recall implementation. In contrast, the Drugs and Cosmetics Act contains Section 26A (prohibition power) and Section 22 (inspector’s powers), but no provision explicitly authorizes the CDSCO to order a recall and mandate return of products from the market. The absence of a statutory recall provision is precisely why the CDSCO’s recall guidelines remain non‑binding – they have no parent section in the Act to give them legal force[43].
Enforcement mechanisms: The FD&C Act provides for civil and criminal penalties, including fines up to $500,000 and imprisonment for knowing violations. The Drugs and Cosmetics Act includes penalties (e.g., Section 27: imprisonment up to life for spurious drugs causing death), but these are focused on manufacturing or selling defective drugs, not on failing to execute a recall. A manufacturer who ignores a recall request may be prosecuted for selling NSQ drugs, but the recall itself is not the subject of the violation. This distinction is important: under the FD&C Act, the refusal to comply with a recall request can be used as evidence of continuing adulteration; under the Drugs and Cosmetics Act, the refusal alone is not a separate offense[44]. Transparency requirements: The FD&C Act, through FDA’s implementing regulations, requires public disclosure of recall information via Weekly Enforcement Reports and the FDA Recall database. The Drugs and Cosmetics Act has no comparable transparency mandate. The CDSCO publishes some recall notices on its website, but there is no legal requirement to do so, and no centralized, searchable database of all drug recalls occurring in India. Synthesis of comparative governance analysis: The US system embodies a “command‑and‑control” model with centralized authority, direct enforcement powers, and statutory codification of recall procedures. The Indian system reflects a “coordinated federal” model where the center sets policy and guidelines, but states execute enforcement. This structural difference means that a recall decision in the US is an actionable order; in India, it is a request that depends on state cooperation and manufacturer goodwill. Consequently, while the CDSCO’s recall guidelines are conceptually sound, their lack of legal force and the fragmented federal governance structure undermine their effectiveness. Any meaningful reform of India’s recall system must address not only the guidelines themselves but also the underlying statutory framework and center‑state implementation mechanisms – an insight that emerges clearly from this comparative governance analysis[45].
CONCLUSION
This comparative study of drug recall procedures in India and the United States reveals fundamental differences in governance structure, statutory authority, and implementation effectiveness, while also highlighting areas of convergence and mutual learning. The US system, built upon decades of legislative refinement following catastrophic events such as the Elixir Sulfanilamide disaster of 1937 and the thalidomide tragedy, represents a mature, centralized model. The FDA operates as a unified federal agency with direct enforcement powers—seizure, injunction, criminal prosecution, and import alerts—that, although not including a standalone mandatory recall order for drugs, create such powerful leverage that voluntary recalls are almost always complied with. The codification of recall procedures in 21 CFR Part 7, the three‑tier classification based on health hazard evaluation, and the statutory requirement for public disclosure through Weekly Enforcement Reports collectively ensure transparency, uniformity, and accountability. Challenges remain, particularly in enforcing recalls against foreign manufacturers and mitigating drug shortages following large withdrawals, but the structural integrity of the US framework is well established.
REFERENCES

Dhriti Biswas, Kriti Dabral, Comparative Study of Drug Recall Procedures in India and the USA, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 355-368. https://doi.org/10.5281/zenodo.20492260
 10.5281/zenodo.20492260
10.5281/zenodo.20492260