
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Appasaheb Birnale College of Pharmacy, South Shivajinagar, Sangli-Miraj Road, Sangli, India 416416
The present study focuses on the computational assessment of new Schiff base compounds based on 1,2,4 pyridine–triazole as possible ligands targeting the GABA-A receptor for anticonvulsant activity. A series of structurally diverse Schiff base analogues containing pyridine and 1,2,4-triazole pharmacophores were designed and investigated using in silico methods to evaluate their binding affinity, pharmacokinetic behaviour, and drug-likeness properties. Molecular docking studies against the GABA-A receptor revealed significant interactions of the synthesized derivatives contains important residues of amino acids within the site that is active, indicating favourable receptor affinity and possible agonistic activity. ADMET prediction and Lipinski’s rule analysis demonstrated that most compounds possess acceptable pharmacokinetic profiles, good oral bioavailability, and low toxicity. Among the evaluated derivatives, certain compounds exhibited stable intermolecular connections and better docking scores in comparison to the standard anticonvulsant drug, suggesting their potential as promising anticonvulsant agents. Furthermore, The aromatic ring's electron-donating and electron-withdrawing substituents were found to significantly influence receptor binding and biological activity. Overall, the study highlights the therapeutic potential of pyridine–triazole based Schiff base scaffolds as novel GABA-A receptor modulators and provides a theoretical foundation for further synthesis and biological evaluation of these compounds for the management of neurological conditions such as epilepsy
About 50 million people worldwide have epilepsy, a long-term neurological disorder marked by frequent convulsions. Despite the widespread availability of antiepileptic medications (AEDs), a considerable percentage of patients still face drug resistance, decreased therapeutic efficacy, and unfavourable side effects. These difficulties have spurred continuous research to create new substances with better therapeutic indices and a stronger capacity to target the nervous system at its core. An imbalance in the brain's excitatory and inhibitory neurotransmission causes epilepsy. One important inhibitory target in the control of seizure activity is the GABA-A (gamma-aminobutyric acid type A) receptor. It is possible to suppress abnormal neuronal discharges and restore normal neurological homeostasis by altering these receptors.(1)
Because of their structural resemblance to the pharmacophores of commonly used antiepileptic medications (AEDs) Derivatives of 1,2,4 pyridine-triazole have shown strong anticonvulsant potential. among heterocyclic compounds. Additionally, by adding more molecular flexibility, the Schiff base moiety enhances pharmacokinetic and pharmacodynamic properties.(2)
The anticonvulsant potential of Schiff base 1,2,4 pyridine-triazole-based compounds are studied in this study using a computer-aided drug design method. Using molecular docking techniques, the binding affinity of fifteen different drugs for the GABA-A receptor (PDB ID: 7DTD) was assessed. Four interesting candidates were further confirmed using in vitro neuronal assays and binding interactions and favourable blood–brain barrier (BBB) permeability.(3)
The combination approach offers a prediction model for finding efficient anticonvulsant drugs with the least number of resources, and the results lay the groundwork for future pharmaceutical research. (See Table No. 1 for Docking scores and complex structures, and Figure No. 1 for molecular interactions.(4)
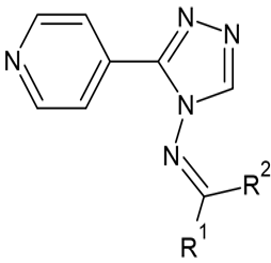
Figure. 1: General structure of selected series of compound 1,2,4 Pyridine Triazole Moiety.
2. MATERIAL AND METHODS:
1. Computational Docking:
1.1) Ligand Preparation: Chem Draw Chem Sketch and Pro 8.0 were used to first sketch the Schiff base derivatives based on 1,2,4 pyridine-triazole. After utilizing PyMOL to transform these 2D structures into 3D conformations, stable geometries appropriate for docking were obtained through energy minimization..(5) After saving the ligands in Auto Dock Tools and PDB format They were converted to PDBQT format using 1.5.7. (6) To enable flexible docking, rotatable bonds were designed and Gasteiger charges were assigned.(7)
1.2) Receptor Preparation: The Protein Data Bank provided the crystal structure of the GABA-A receptor (PDB ID:7DTD).(8) Making Use of Tools for Auto Dock 1.5.7, polar hydrogens were added, water molecules were eliminated, and the receptor's Gasteiger and Kollman charges were calculated.(9) To provide sufficient ligand interaction, To hold the leftovers from the active site, a grid box was constructed. For application in the docking simulation, the receptor was stored in PDBQT format. (10)
1.3) Validation of Docking Protocol: To confirm the docking process, the co-crystallized ligand from the GABA-A receptor structure was re-docked into its binding site using the same grid and docking parameters. (11) The root-mean-square deviation (RMSD) of the docked pose) from the initial ligand position was computed. The precision and dependability of the docking configuration were verified by an RMSD value of 0.821 Å (less than 2.0 Å). (12) To ensure that the docking data were biologically relevant, The Visualizer for Biovia Discovery Studio 2024 was also used to evaluate and validate important chemical interactions, including as hydrophobic contacts and hydrogen bonding. (13)
2. Drug likeness studies and ADME prediction: For each of the proposed compounds (D1, D2, D3, and D4), molecular properties and drug-likeness parameters were computed. The drug-likeness study is important because it identifies molecules that fit the definition of druglike molecules. Research on ADME attributes is crucial because of the pharmacokinetic properties of pharmacological compounds, their oral bioavailability, cell penetration, metabolism, and elimination. Using SWISS ADME techniques and other physicochemical variables like molecule refractivity, Lipinski's rule of five was calculated Water solubility and GI absorption The number of rotatable bonds and the journal were expected to This study used a number of software tools to ensure accurate forecasts and comprehensive analysis. SWISS ADME was used to predict absorption, distribution, metabolism, and excretion (ADME), which provided crucial details about the drug's pharmacokinetic properties. ProTox 3.0 was used to assess the toxicity profiles of the compounds and ensure their safety for further investigation. (30)
3. Boiled EGG analysis: The gastrointestinal absorption and blood–brain barrier permeability of the selected substances was evaluated using the BOILED-Egg model. The study found that drugs in the white region were linked to higher intestine absorption capability, while those in the yellow zone were expected to have increased permeability across the blood–brain barrier. The Swiss ADME digital platform was used to conduct the study. (31)
4. Toxicity studies: To evaluate the safety profile of the synthesized chemicals, a toxicology investigation was conducted. Acute toxicity (LD⋅₀), toxicity class, and organ-specific toxicities including hepatotoxicity, mutagenicity, and carcinogenicity were estimated using ProTox-II (version 3.0), an in silico prediction platform. It uses machine learning and molecular similarity techniques to enable effective early-stage screening of possible hazardous effects based on SMILES input.
5. Anticonvulsant Activity: A substance's pharmacological capacity to stop or lessen the frequency and severity of seizures is referred to as anticonvulsant activity. Epileptic seizures arise due to abnormal and excessive The brain's neuronal discharge is frequently linked to an imbalance between excitatory and inhibitory neurotransmission. Conventional anticonvulsant drugs primarily act by enhancing γ-aminobutyric acid (GABA)-mediated inhibitory pathways, blocking voltage-gated sodium or calcium channels, or reducing excitatory glutamatergic transmission in the central nervous system. Clinically used antiepileptic drugs such as phenytoin, carbamazepine, valproic acid, and benzodiazepines exert their effects through modulation of ion channels or neurotransmitter systems. However, many currently available anticonvulsants are associated with adverse effects including sedation, dizziness, cognitive impairment, tolerance, and hepatotoxicity, while some patients continue to experience drug-resistant epilepsy. Consequently, recent research has focused on identifying novel molecular targets and safer therapeutic agents with improved efficacy and reduced side effects. Emerging targets include GABA-A receptor subtypes, NMDA receptors, voltage-gated ion channels, and neuroinflammatory pathways involved in seizure generation and propagation. Anticonvulsant activity is commonly evaluated using a combination of in vitro assays, molecular docking investigations conducted in silico and in vivo experimental models like the Pentylenetetrazol (PTZ)-induced seizure model, which help in assessing the seizure-protective potential of newly synthesized compounds. (32)
3. RESULTS AND DISCUSSION:
Table 1: Docking Scores of 1,2,4 Pyridine Triazole Schiff Base Derivatives with GABA-A Receptor.
|
Sr. No |
Compound Name |
DR1R2 |
Binding Score (Kcal/Mol) |
|
|
D1 |
4-Bromoacetophenone |
-6.4 |
|
|
D2 |
4-Chloroacetophenone |
-8.2 |
|
|
D3 |
4-Hydroxyacetophenone |
-7.8 |
|
|
D4 |
4-Methyleacetophenone |
-7.8 |
|
|
D5 |
4-Nitroacetophenone |
-6.5 |
|
|
D6 |
Acetophenone |
-8.1 |
|
|
D7 |
Benzophenone |
-9.0 |
|
|
D8 |
Butanone |
-7.3 |
|
|
D9 |
Cyclohexanone |
-7.1 |
|
|
D10 |
Ethyl methyl ketone |
-6.4 |
|
|
D11 |
Methyl Propyl Ketone |
-7.2 |
|
|
D12 |
Methylcyclohexanone |
-6.9 |
|
|
D13 |
Pentane-3-one |
-7.7 |
|
|
D14 |
Hexane-2-one |
-7.3 |
|
|
D15 |
propiophenone |
-8.4 |
Table 2: The compounds that can cross the BBB are chosen to synthesise various derivatives of Benzophenone, 4-Hydroxyacetophenone, Cyclohexanone, 4-Bromoacetophenone. (D7, D3, D9, D1)
|
Sr. No |
Compounds |
Anticonvulsant Docking score |
|
1. |
D7 |
-9.0 |
|
2. |
D3 |
-7.8 |
|
3. |
D9 |
-7.1 |
|
4. |
D1 |
-6.4 |
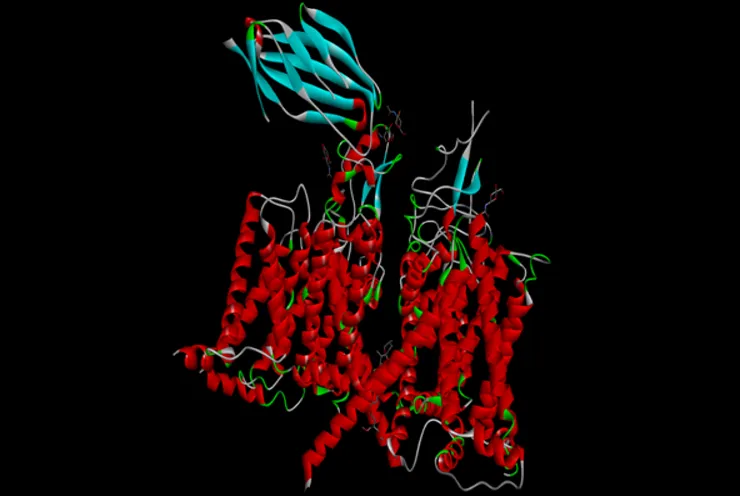
Figure No 2: PDBID: 7DTD
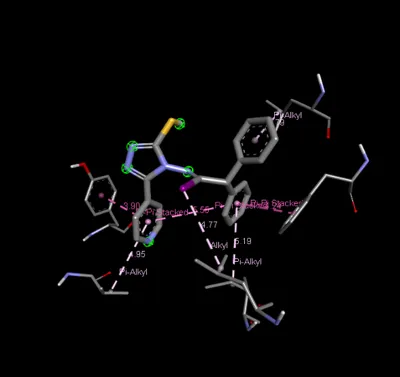
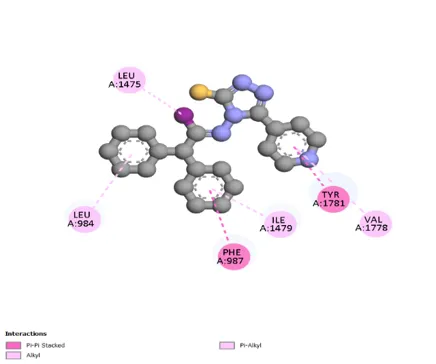
Figure 3: 2D & 3D Representation of Docking interaction with receptor of compound Benzophenone (D7) with PDBID: 7DTD
Table 3: Docking interaction of receptor (7DTD) with compound Benzophenone (D7)
|
Sr. No. |
Residue Atom |
Distance |
Category |
Type of Interaction |
|
1 |
A: LEU 1475 |
4.77 |
Hydrophobic |
Pi-Pi Stacked |
|
2 |
A: LEU 984 |
4.79 |
Hydrophobic |
Alkyl |
|
3 |
A: PHE 987 |
4.23633 |
Hydrophobic |
Pi-Pai Stacked |
|
4 |
A: ILE 1479 |
5.19 |
Hydrophobic |
Pi Alkyl |
|
5 |
A: TYR 1781 |
3.89632 |
Hydrophobic |
Pi-Pi Stacked |
|
6 |
A: VAL 1778 |
4.95 |
Hydrophobic |
Alkyl |
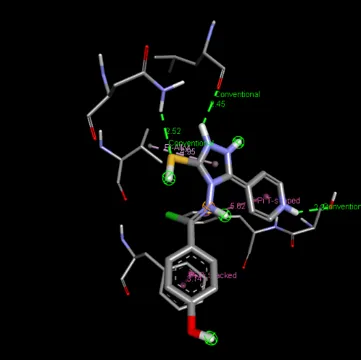
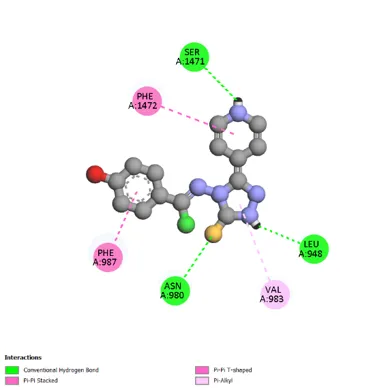
Figure 4: 2D & 3D Representation of Docking interaction with receptor of compound 4-Hydroxyactophenone (D3) with PDBID: 7DTD
Table 4: Docking interaction of receptor (7DTD) with compound 4-Hydroxyacetophenone (D3)
|
Sr. No. |
Residue Atom |
Distance |
Category |
Type of Interaction |
|
1 |
A: SER 1471 |
2.34 |
Hydrophobic |
Conventional Hydrogen Bond |
|
2 |
A: PHE 1472 |
5.62 |
Hydrophobic |
Pi-Pi shaped |
|
3 |
A: PHE 987 |
3.74 |
Hydrophobic |
Pi-Pi Stacked |
|
4 |
A: ASN 980 |
2.52 |
Hydrophobic |
Conventional Hydrogen Bond |
|
5 |
A: VAL 983 |
4.95 |
Hydrophobic |
Conventional Hydrogen Bond |
|
6 |
A: LEU 948 |
2.45 |
Hydrophobic |
Pi-Alkyl |
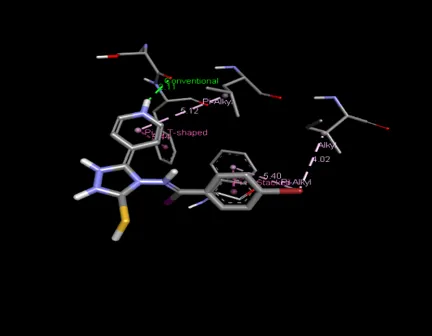
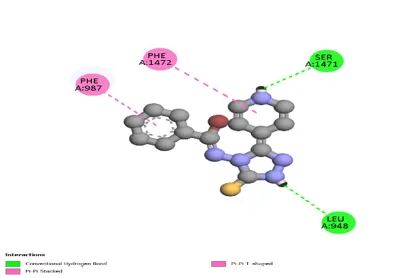
Figure 5: Docking interaction with receptor of compound Cyclohexanone (D9) with PDBID: 7DTD
Table 5: Docking interaction of receptor (7DTD) with compound Cyclohexanone (D9)
|
Sr. No. |
Residue Atom |
Distance |
Category |
Type of Interaction |
|
1 |
A: SER 1471 |
2.59 |
Hydrophobic |
Conventional Hydrogen Bond |
|
2 |
A: PHE 1472 |
5.55 |
Hydrophobic |
Pi-Pi shaped |
|
3 |
A: PHE 987 |
3.78 |
Hydrophobic |
Pi-Pi Stacked |
|
4 |
A: ASN 948 |
2.89 |
Hydrophobic |
Conventional Hydrogen Bond |
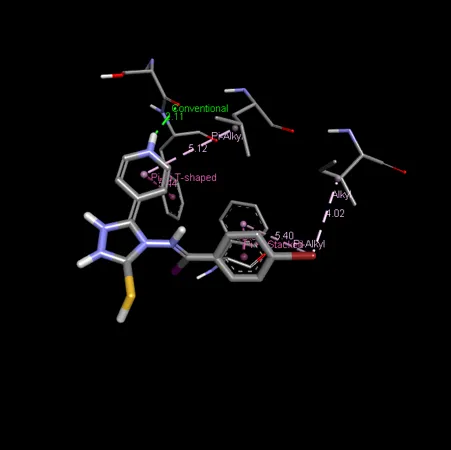
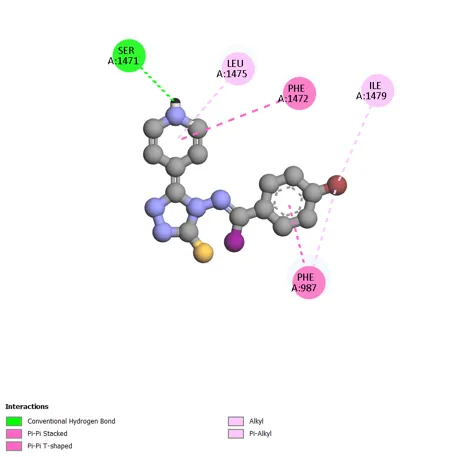
Figure 6: 2D & 3D Representation of Docking interaction with receptor of compound 4-Bromoacetophenone (D1) with PDBID: 7DTD
Table 6: Docking interaction of receptor (7DTD) with compound 4-Bromoacetophenone (D1)
|
Sr. No. |
Residue Atom |
Distance |
Category |
Type of Interaction |
|
1 |
A: SER 1471 |
2.11 |
Hydrophobic |
Conventional Hydrogen Bond |
|
2 |
A: LEU1475 |
5.12 |
Hydrophobic |
Alkyl |
|
3 |
A: PHE 1442 |
5.44 |
Hydrophobic |
Pi-Pi Stacked |
|
4 |
A: ILE 1479 |
4.02 |
Hydrophobic |
Pi-Alkyl |
|
5 |
A: PHE 987 |
3.96 |
Hydrophobic |
Pi-Pi Stacked |
Table 7: Screening of Drug-likeness Parameters
|
Sr. No. |
Ligand |
MF |
Lipinski’s Rule of 5 |
Lipinski’s Violation |
Lipinski’s Rule |
|||
|
MW (g/mol) |
Log P |
HBA |
HBD |
|||||
|
1 |
D1 |
C21H16IN5S |
497.36 |
5.05 |
6 |
1 |
0 |
Yes |
|
2 |
D2 |
C16H14CIN5S |
343.82 |
3.82 |
6 |
1 |
0 |
Yes |
|
3 |
D3 |
C14H9BRIN5S |
390.21 |
3.12 |
4 |
2 |
0 |
Yes |
|
4 |
D4 |
C14H10CIN5OS |
486.13 |
3.01 |
4 |
1 |
0 |
Yes |
Table 8: Evaluation Of In-Silico ADMET Parameters
|
Ligand |
In-silico ADMET |
|
|
|||||
|
Absorption |
Distribution |
Metabolism |
|
Toxicity |
||||
|
Water Solubility |
BBB permeability |
1A2 |
2C19 |
2C9 |
2D6 |
3A4 |
Predicted LD50 & Toxicity Class: |
|
|
D1 |
-6.70 (Moderately soluble) |
3.55 |
Yes |
Yes |
Yes |
No |
No |
1190mg/kg (Class:4) |
|
D2 |
-5.47 (Moderately Soluble) |
2.75 |
Yes |
Yes |
Yes |
No |
No |
1190mg/kg (Class:4) |
|
D3 |
-5.72 (Moderately soluble) |
3.25 |
Yes |
Yes |
Yes |
No |
No |
1190mg/kg Class: 4 |
|
D4 |
-5.01 (Moderately Soluble) |
2.82 |
Yes |
No |
No |
No |
No |
1190mg/kg Class: 4 |
4. CONCLUSION
The present computational study demonstrated that 1,2,4 pyridine–triazole based Schiff base derivatives four derivatives D1, D2, D3 and D4 possess promising affinity toward the GABA-A receptor and may serve as potential anticonvulsant agents. Molecular docking analysis revealed favourable binding interactions of the synthesized derivatives with significant amino acid residues found in the receptor's active site, indicating their potential modulatory activity. The evaluated compounds also exhibited satisfactory pharmacokinetic and drug-likeness characteristics based on Lipinski's rule analysis and ADMET, suggesting acceptable bioavailability and safety profiles. Structural modifications involving electron-donating and electron-withdrawing substituents were found to significantly influence receptor binding efficiency and stability of ligand–receptor interactions. Among the investigated derivatives, selected compounds displayed better docking scores and interaction patterns comparable to standard anticonvulsant drugs, highlighting their potential as lead molecules for further development. Overall, the findings of this study provide valuable theoretical insight into the design of novel pyridine–triazole Schiff base derivatives and support its additional production, in vitro testing, and in vivo assessment to create therapeutic compounds that effectively treat epilepsy and other neurological disorders.
REFERENCES

Vaibhav Patil, Dr. Bhavana Jain, Dr. Manish Kondwar, Computational Evaluation of 1,2,4 Pyridine-Triazole Based Schiff Base Derivatives Targeting the Gaba-A Receptor, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 2087-2096. https://doi.org/10.5281/zenodo.20595524
 10.5281/zenodo.20595524
10.5281/zenodo.20595524