
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1,2IT college Department of Biotechnology, Lucknow
3SGPGIMS Lucknow
*Isabella Thoburn College Of Professional Studies, Lucknow
The increasing number of people suffering from neurological disorders is concerning and hence, there is an immediate need to build an effective therapy to lessen this burden. For the past decade, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated protein (Cas) (CRISPR/Cas) system has been gaining popularity as a gene editing tool for numerous genetic disorders due to its simplicity, easy-to-use, easy access to researchers and most importantly its ability to target multiple genes at a time irrespective of the cell type. Limited knowledge about the pathophysiology of complex neurological disorders, restricted access to the brain due to the blood brain barrier and off-target effects of CRISPR/Cas are major areas of concerns. Nevertheless, researchers have demonstrated the potential of CRISPR/Cas gene editing and the promise it holds as a therapeutic approach against central nervous system disorders.
The pace of inability and mortality of neurological and neuropsychiatric turmoil is essentially higher in contrast with other human diseases. Therefore, more consideration is expected in the early determination of these human problems [1]. Genetic disease is an inevitable by-product of evolution. Indeed, evolutionary stress has increased the likelihood of developing many neuropsychiatric or neurodevelopmental diseases [2]. Neurological disease may occur as a result of a tumor, degeneration, infections, damage in a structure, or trauma or may be conditioned by birth i.e., are congenital and are marked by a range of symptoms including seizures, loss of sense, pain, paralysis, weak muscles, etc. [3]. For over three decades, many scientists have perpetuated that hereditary changes would give successful therapy for many human diseases that are heritable, offering sturdy and potentially therapeutic clinical advantages with a solitary treatment [4].
While many neural activities' molecular underpinnings are still poorly understood, developments in next-generation sequencing (NGS) and technologies involving gene editing have started to fill this void. A highly effective gene manipulating tool termed a Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated protein (Cas) framework has surfaced and is capable of altering the genome irrespective of the living being and cell type [5]. Because of its modularity, precision, and accessibility, the recent remodeling of this prokaryotic immune system CRISPR for eukaryotic genome editing has revived the subject [6]. The capacity to modify the type of genetic regulation allows a gene therapy method like CRISPR and other related technologies to potentially be used for a wide range of therapeutic implications within and outside of the central nervous system (CNS) [7].
This review provides ideas on therapeutic gene editing for neurological diseases by summarizing recent investigations of CRISPR/Cas applications in models of neurological disorders. Basic mechanism of CRISPR/Cas9 and its delivery methods have also been discussed.
2. CRISPR: AS A GENE EDITING TOOL
The area of gene therapy, requiring genome editing as a technique to rectify the genetic abnormalities that cause disease, became particularly focused on the need to employ tools to build treatments [8]. DNA is changed at the single-base level during genome editing, a form of genetic engineering. The area of biomedical research is being metamorphosed by genome editing, which has the potential to treat or prevent a wide range of human genetic problems [9]. The periodic suppression of DNA double-strand break (DSB) repair is unquestionably the most striking illustration of DNA repair regulation by the cell cycle. Non-homologous end joining (NHEJ) and homologous recombination (HR), two key kinds of repair processes, are used to fix these DSBs [10]. A DNA template with ends that are homologous to the break site is provided and used to copy the information across the break in highly accurate homology-directed repair (HDR), as opposed to the non-homologous end-joining (NHEJ) pathway, which stitches two ends of the break back together with indels (insertion or deletion) of nucleotides. Functional genomic research can benefit greatly from NHEJ repair because it is highly useful in producing functional gene knockouts. HDR, on the other hand, is primarily utilized in genome editing to modify the DNA sequence and produce new versions of genes or proteins [11].
There are four key techniques, viz. meganucleases (MegNs), transcription activator-like effector nuclease (TALENs), zinc finger nucleases (ZFNs), and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) (CRISPR/Cas-9) [12]. of site-specific genome editing that has cleared the way for new medicinal and agricultural discoveries in the field of targeted nucleases and their prospective application to model and non-model species [12]. The introduction of the CRISPR/Cas9 technology has given gene therapy a window of opportunity to shed its negative connotations and establish itself as an effective therapeutic approach [13]. The CRISPR-Cas system, which confers adaptive protection in many bacteria and most archaea, is made up of clustered regularly interspaced short palindromic repeats (CRISPR) and related proteins (Cas) [14]. The short, partially palindromic DNA repeats found in CRISPR loci are spaced regularly, forming loci that alternate repeating elements (CRISPR repeats) with varied sequences (CRISPR spacers). The size of CRISPR spacers can also vary (21-72 nt), but they are normally 32–38 nt in length. CRISPR repeats can vary greatly (23–55 nt), and they are typically 28–37 nt long [15].
CRISPR was identified for the first time in the genome of Escherichia coli in 1987 by Japanese researchers as a collection of short direct repeats interspersed with short sequences [16]. It was proposed that antisense RNAs may be used by CRISPR-Cas as an adaptive defense system that can remember the hallmarks of previous invasions in conjunction with the discovery that CRISPR loci are transcribed and the observation that cas (CRISPR-associated) genes encode proteins with supposed nuclease and helicase domains [17]. Given their genetic makeup and structural and functional variations, CRISPR-Cas systems have been divided into three major categories, referred to as type I, type II, and type III, and a total of 12 subtypes [18]. The cas genes and the proteins they express are the primary characteristics that distinguish CRISPR-Cas types and subtypes; they are very genetically and functionally variegated, illuminating the myriad biochemical tasks that they carry all around the several steps of CRISPR-mediated adaptive immunity. Immune systems using CRISPR-Cas generally work in three phases, (i) Adaptation: additional spacers are incorporated into the CRISPR locus from external nucleic acid, (ii) crRNA biogenesis: which involves the transcription and processing of CRISPR exhibits into tiny interfering CRISPR RNAs (crRNAs) and, (iii) Targeting: homologous sequences are cleaved specifically by cas nucleases which are led by these crRNAs to the target site [18].
Streptococcus thermophilus harbored a big gene that encoded a single-effector protein with nuclease activity known as Cas9, also discovered the protospacer adjacent motif (PAM), which is a common sequence in the target DNA next to the spacer and is required for Cas9 to recognize and bind its target DNA spacers are added to the CRISPR RNAs (crRNAs), which direct the Cas proteins to the DNA target site. PAMs signal “non-self” / foreign strands when they are present, whereas they signal "self" when they are absent. Pam sequences vary amongst Cas9 orthologs from various species [19]. Trans-activating crRNA (tracrRNA), a short RNA that complements the repetition sections of crRNA, is also necessary for Cas9 to function [20].
The CRISPR/Cas9 system uses a truncated duplex RNA that is joined together to produce a stem-loop structure that can bind to the target site [21]. Cas9 can precisely knock out or knock in target genes by directing sgRNA, which is made up of CRISPR-derived RNA and trans-activating CRISPR RNA (tracrRNA), which together comprise the two -RNA structure of sgRNA [22]. The structure of the sgRNA is straight and straightforward, making it simple to build. Nearly every place of the genome where the complementary sgRNA is chosen can be connected to the Cas9-based structure with relative ease. Apparently, the cas9 nuclease cleaves 3 base pairs upstream PAM site (5’-NGG-3’) in the target gene. The PAM sequence is preceded by a 20 base pairs long DNA sequence which the CRISPR/cas9 system aims at. This results in DSBs that can occur through either homologous recombination (HR) or nonhomologous end-joining (NHEJ) [23] (Figure 1). Inactivation of both HNH and RuvC domains changes CRISPR/Cas9 system in such a way that it will function as an effective DNA binding tool but will no longer have endonuclease activity. Such CRISPR/Cas9 is termed as deactivated/dead Cas9 (dCas9). This mechanism can be combined with a variety of modifiers, such as epigenetic effectors, to modulate gene expression [24].
The CRISPR/Cas9 system's specificity is expressed by the fact that the genomic modifications it introduces are restricted to the precise target spot and are far from the off-target sequence [25]. Therefore, by simply altering the small portion of the guide RNA that determines specificity, which can be accomplished in a single cloning process, different Cas proteins can be directed to different DNA sequences [26].
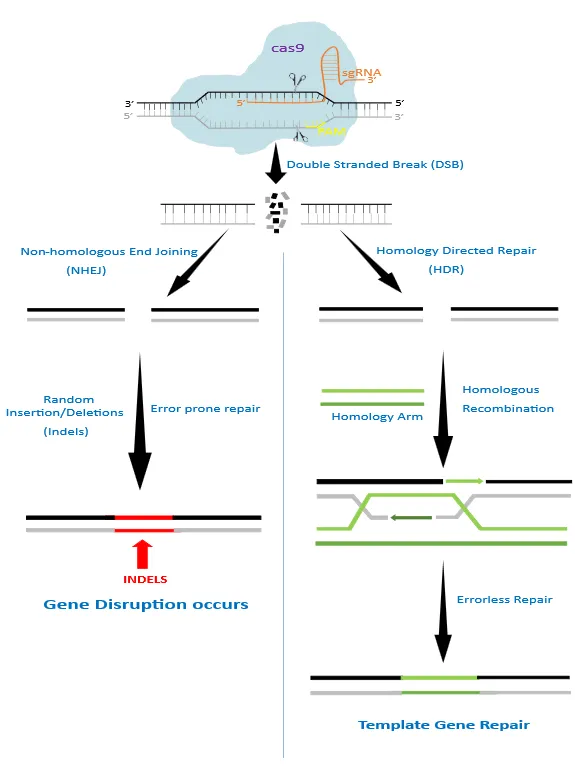
Figure 1: Mechanism of CRISPR/Cas9 gene editing. Adapted from [13]
3. Delivering Crispr To The Brain
The blood-brain barrier (BBB), which serves the purpose of segregating and protecting neural tissue while regulating molecule access and so impeding delivery, presents the primary challenge when developing a therapeutic for the brain [27]. Since CRISPR has a weak capacity for cell entry and is unstable and biodegradable, appropriate delivery techniques to the target cells or organs are therefore required to solve these issues. The three primary delivery modalities for the CRISPR/Cas9 system are physical, viral, and non-viral [28].
3.1. Physical method –
3.1.1 Microinjection
When compared to the virus carrier delivery technology, microinjection has an advantage in that it has a huge loading capacity and is unrestricted. It also allows for precise injection into predetermined areas rather than into a complex natural milieu within the body, making it ideal for in vitro research [28].
3.1.2 Electroporation uses brief electrical current pulses to trigger the temporary opening of cell membrane pores, allowing payloads to enter cells and since electroporation effectively transports payloads into a wide range of cell types, it is frequently utilized in ex vivo and in vitro gene editing [28].
3.1.3 Hydrodynamic delivery is an in vivo administration approach that is quick, effective, and secure. Syringes can be used to quickly inject medications into veins, allowing them to act in vivo and treat diseases [28].
3.2. Viral Vectors -
3.2.1 Adeno-associated virus (AAV) It is an encapsulated single-stranded DNA virus. AAVs can introduce CRISPR/Cas9 in one of two ways: (i), AAVs can be used as a transductional delivery system for Cas9, sgRNAs, and/or donor templates. (ii), the HDR route allows for the use of AAV vectors as a donor template for gene knock-in [29]. AAV vectors' negligible cloning capacity (less than 4.7 kb) is what restricts them [30].
3.2.2 Lentiviruses (LVs) The ability to clone both Cas9 and sgRNA into a single LV vector is made possible by the larger cloning capacity (less than 8 kb) of LV vectors compared to AAV vectors. Compared to AAV manufacture, LV production is also less irksome. However, the major issue with LV systems is their unpredictable incorporation into host cell genomes [30]. To avoid the possibility of insertional mutagenesis in the brain, integration-defective lentiviral vectors have also been designed [31].
3.2.3 Adenoviruses (AVs) AVs can infect both proliferating and nondividing cells and, most significantly, do not integrate into the genomes of the hosts [32]. The main difficulty with using AVs for distribution is that they cause strong innate immunological reactions in host cells, which leads to tissue inflammation and the eventual elimination of AV vectors. The applicability and effectiveness of this method are constrained by the labor-intensive nature of AV production [30].
3.3. Non-viral vectors –
3.3.1 Liposomes They are made up of spherical, concentric bilipid layers that can transport nucleic acids to their intended target cells. Larger nucleic acids can be transfected more effectively and conveniently with cationic liposomes. Trojan horse liposomes (THL) have been produced to promote BBB crossing [31]. To bind with the appropriate receptors, such as transferrin receptors or insulin receptors, that are located on the BBB or the cell surface, THL depends on the monoclonal antibody component of the compound [27].
3.3.2 Polymer-based vectors: Cationic polymers like poly-L-lysine (PLL) and polyethyleneimine (PEI) are frequently investigated in vivo in the CNS for gene therapy. PLL is special because of its biodegradable nature, which is advantageous for in vivo use; PEI is handy since it can be made to be varied lengths, be branching or straight, and undergo functional group replacement or addition [27].
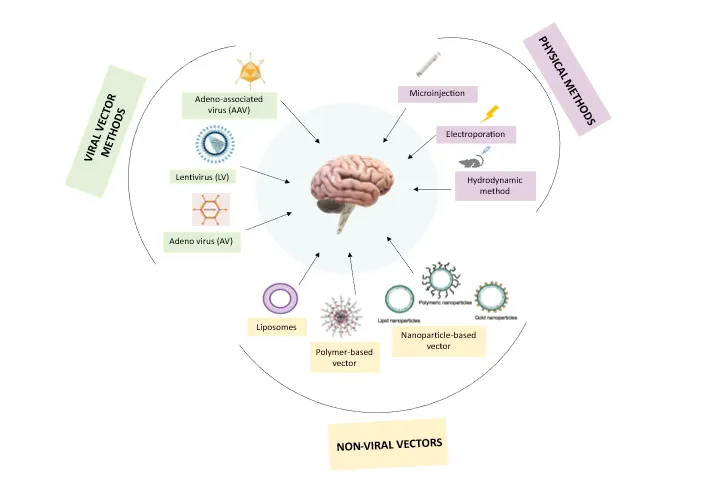
Figure 2: Delivery methods of CRISPR
3.3.3 Nanoparticle-based vectors Nanoparticles are solid colloidal particles that range in size from 1 to 1000 nm and are made of macromolecular substances [33]. The active ingredient (a medication or physiologically active component like DNA, RNA, or protein) is encapsulated, absorbed, or entrapped within these particles. Poly-butylcyanoacrylide (PBCA), poly-lactic acid (PLA), and similar copolymers are important constituents of nanoparticles. The general biocompatibility and quick biodegradability of nanoparticles make them attractive gene delivery agents for CNS targeting [27] (Figure 2).
4. WHY CRISPR?
The ability to alter the majority of biological processes by focusing on the underlying genes is one of the main benefits of gene therapy. Focusing on the underlying pathologic processes can slow or stop the progression of the disease, in contrast to traditional medicine, which frequently concentrates on symptom treatment. The ability to treat diseases with a single administration may also be enabled by steady transgenic expression or long-lasting genome alteration [34]. With zinc fingers (ZFs) and transcription activator-like effector nucleases (TALENs), the first attempts at targeted gene editing were carried out [35]. Both of these nucleases rely on programmable DNA-binding proteins connected to active endonucleases to break particular DNA regions. Despite being appropriate for a wide range of applications, these techniques have lost favor with emerging genome editing systems as a result of relative drawbacks such as their demanding requirements for protein engineering. Clustered regularly interspaced palindromic repeats (CRISPR)-Cas9 is the result of recent developments in gene editing technology. A programmable single guide RNA (sgRNA) is used by CRISPR nucleases to target complementary DNA sequences for cleavage, in contrast to its forerunners [5][36]. Cas9 can facilitate targeted DNA DSBs at particular loci of interest in the mammalian genome and promote genome editing via NHEJ or HDR. Over ZFNs and TALENs, Cas9 has a number of benefits, such as easier customization, better-targeting accuracy, and support for multiplex genome editing. By simply acquiring a pair of oligos that encode the 20-nt guide sequence, Cas9 may be easily retargeted to fresh DNA sequences [36]. The ease of use and broad application of the CRISPR-Cas9 system in genome alterations of practically all biological systems examined so far, including stem cells, cell lines, yeasts, worms, insects, rodents, and mammals, are its most significant advantages. Besides genome editing, dCas9 (dead Cas9) has also been used to alter entire genome gene expression profiles by together activating (CRISPRa) or inhibiting (CRISPRi) the expression of one or more than one target genes [37].
The foundation of ZFNs and TALENs is protein-guided DNA cleavage, which calls for labor-intensive and intricate protein engineering, selection, and validation. For DNA targeting, CRISPR/Cas9 only requires a brief programmable gRNA, which is relatively inexpensive and simple to design and create. CRISPR/Cas9 is able to simultaneously induce genomic changes at a number of different independent sites by utilizing Cas9 and a number of gRNA with diverse target sites [38]. With the use of this technology, it is possible to produce transgenic animals more quickly with a variety of gene mutations and to disrupt a number of genes or an entire gene family in order to study gene function and epistatic connections [38]. CRISPR-Cas technologies have the potential to revolutionize basic and translational brain research through precise and effective gene editing [26].
5. Crispr-Mediated Treatment Approach In Neurological Disorders
5.1 Alzheimer’s disease
Extracellular senile plaques are a key pathogenic aspect of Alzheimer's disease (AD), that is seen in the brain. Aggregations of β-amyloid (Aβ), a tiny peptide, make up senile plaques. Multiple lines of evidence show that Aβ overproduction or aggregation in the brain is a major contributing factor to AD, and inhibition of Aβ synthesis has gained significant attention in AD research [39].
In a recent study, levels of CysLT1R in AD patients and APP/PS1 mice were examined. Additionally, by using the CRISPR/CRISPR-associated protein 9 (Cas9) system to delete the CysLT1R gene in APP/PS1 mice, researchers were able to produce APP/PS1-CysLT1R-/- mice. These mice were then used to study the effects of CysLT1R knockout on amyloidogenesis, neuroinflammation, the structure of synapse and its plasticity, cognition, and the kynurenine pathway. After the CysLT1R gene in APP/PS1 mice was knocked down through lentivirus, these stated characteristics were also investigated. They discovered that in APP/PS1 mice, CysLT1R deletion or knockdown could preserve synaptic structure and plasticity and enhance cognition. These benefits were linked to concomitant reductions in neuroinflammation, amyloid processing, and the kynurenine pathway. Therefore, the researchers concluded that CysLT1R deficiency can have a number of protective benefits against AD development, and genetic or pharmaceutical ablation of this protein may be an AD treatment option [40].
Numerous neurodegenerative diseases, including Alzheimer's disease (AD), have elevated levels of microglia-specific genetic variations, suggesting that changes to the innate immune system play a significant role in the disease's etiology. In order to evaluate the protective variant's role in the context of immune cell functions, researchers used CRISPR/Cas9 gene editing to create a Plc2-P522R knock-in (KI) mouse model. This revealed an atypical coding variant in the PLCG2 gene (rs72824905, p.P522R) expressed in myeloid lineage cells and appears to lower the risk for AD [41].
Park et al., (2019) in an experiment involving five familial Alzheimer’s disease (5XFAD) and amyloid precursor protein (APP) knock-in Alzheimer’s disease mouse models successfully demonstrated the therapeutic application of CRISPR/Cas9. Using “CRISPR-Cas9-loaded” nanocomplexes (developed by adding amphiphilic R7L10 peptide to Cas9-sgRNA), the team, effectively targeted the Bace1 gene in the post-mitotic neuron in the brain of an adult mouse. They then examined the ability of Cas9 nanocomplexes targeting Bace1 to reduce Aβ levels and treat memory loss in Alzheimer's disease models. It was detected that effective Bace1 targeting in the brains of mice that had received injections of the nanocomplex, and as a result, there was a roughly 70% reduction in Bace1 expression in the CA3 region of the treated animals compared to the untreated mice. There was also a significant decrease in β-cleavage products of amyloid precursor protein (APP) in treated 5XFAD brains. The findings show that Cas9 nanocomplex-mediated targeting of Bace1 improves Aβ-associated pathologies and cognitive loss in 5XFAD as well as in App knock-in mice models of Alzheimer's disease [42].
Although some pathogenic variants for Alzheimer's disease (AD) have been discovered, protective mutations have also been discovered in humans. A potential protective deletion mutation was identified in 3′-UTR of the gene encoding the amyloid precursor protein (App) in mice. The study involved a knock-in mouse model for the endogenous App gene that has three AD mutations and a humanized Aβ sequence. Animals with different App 3′-UTR deletions and genetic mosaicism are created when the model zygotes' genomes are edited utilizing a variety of CRISPR/Cas9 tool combinations. Based on the editing quality, the 3′-UTR disruption slows the progression of Aβ disease by controlling APP expression at the transcriptional and translational levels. Remarkably, Aβ pathology is significantly lessened in an App knock-in mouse with a 34-bp deletion in a 52-bp regulatory region next to the stop codon [43]. A new tau knockout strain (tauΔex1) that was created by altering the Mapt gene's intron -1/exon 1 in C57Bl/6J mice was reported by Tan et al., (2018). Although tauΔex1 lacked an obvious phenotype, they had a markedly decreased vulnerability to excitotoxic convulsions and young mice's memory formation was unaffected [44].
5.2 Parkinson’s Disease
A heterogeneous neurological disorder called Parkinson's disease (PD) approximately affects 10 million people worldwide. Motor symptoms like rest tremors, bradykinesia, and stiffness are the mainstays of PD clinical features and are brought on by the progressive death of dopaminergic neurons in the substantia nigra pars compacta (SNpc). Lewy bodies (LBs), which are predominantly formed of clumps of misfolded α-synuclein protein and may spread in a prion-like manner between synaptically linked locations, develop after this neuronal loss [45]. Parkinson's disease (PD) is mostly caused by mutations in the proteins α-synuclein (SNCA), PTEN-induced putative kinase 1 (PINK1), Parkin RBR E3 Ubiquitin Protein Ligase (PARK2), DJ-1 (PARK7), and leucine-rich repeat kinase 2 (LRRK2) [46]. A handful of studies have been conducted indicating the effectiveness of CRISPR technology as a therapeutic option for PD.
A recent study, in the striatum of 6-OHDA-lesioned rats, activated the endogenous tyrosine hydroxylase gene (th) of astrocytes to create dopamine (DA) as a proof-of-concept for the treatment of Parkinson's disease (PD) using SAM, a CRISPR gene activation system. The expression of the Th protein in the C6 glial cell line was assessed, and putative sgRNAs within the rat th promoter region were examined. The SAM complex and the chosen sgRNA were introduced into rat astrocyte cultures using pseudo-lentivirus, and gene expression and Th protein synthesis were measured; also, the release of DA into the culture medium was assessed. Compared to lesioned rats receiving astrocytes that did not produce DA, improvement in motor behaviour in the lesioned rats who got DA-astrocytes was observed. The data demonstrate that DA-producing astrocytes can be produced by SAM-induced expression of the astrocyte's endogenous th gene, which effectively lessens the motor asymmetry brought on by the lesion [47].
Another study conducted by Sastre et al., (2023) showed in iPSC-derived neuronal cells generated from a patient with an SNCA genetic triplication, CRISPRi downregulation of alpha-synuclein demonstrated functional recovery through decrease of mitochondrial DNA damage and oxidative stress. They constructed a CRISPR interference (CRISPRi) system based on the nuclease-dead S. aureus Cas9 (SadCas9) fused with the transcriptional repressor domain Krueppel-associated box to precisely address alpha-synuclein gene regulation. After screening single guide (sg)RNAs across the SNCA promoter, their team located multiple sgRNAs that, at different levels, cause alpha-synuclein downregulation. These findings demonstrate the viability of precision medicine in vitro by focusing on the SNCA gene promoter, thereby providing a potential therapeutic option for PD [48].
Yoon et al., (2022), both in vitro and in vivo, deleted A53T-SNCA using the CRISPR-Cas9 method. In vitro, A53T-SNCA expression levels were markedly decreased by Adeno-associated viruses harboring SaCas9 (Staphylococcus aureus)-KKH with a single guide RNA targeting A53T-SNCA. Using a viral A53T-SNCA-overexpressing rat model of PD, they evaluated its therapeutic potential in vivo. The upregulation of α-synuclein, dopaminergic neurodegeneration, reactive microgliosis, and parkinsonian motor symptoms were all dramatically reversed by gene deletion of A53T-SNCA [49].
Another study by Chen et al., (2019) showed that CRISPR/Cas9n-mediated gene editing reduces or totally eliminates SNCA alleles, conferring a degree of resistance to Lewy disease. They created SNCA+/- and SNCA-/- cell lines by using CRISPR/Cas9n technology to remove the endogenous SNCA gene, which codes for the protein α-synuclein, in a clinical-grade hESC (human embryonic stem cells) line. Researchers first transformed hESC lines into mDA (midbrain dopaminergic) neurons, and after that, these were exposed to recombinant α-synuclein preformed fibrils (PFFs) to stimulate the development of Lewy-like disease as shown by the phosphorylation of serine-129 of α-synuclein (pS129- αSyn). While SNCA+/- and SNCA-/- neurons showed significant resistance to the production of this pathogenic mark, wild-type neurons were completely susceptible to the creation of protein aggregates positive for pS129-Syn [50].
SNCA intron 1 DNA methylation controls SNCA transcription, and PD brains have differing levels of DNA methylation compared to controls. Thus, for precise downregulation of SNCA levels, DNA methylation at SNCA intron 1 is a desirable target. Kantor et al., (2018) created a technique for precise DNA methylation editing within intron 1 using an “all-in-one” lentiviral vector. The system is composed of CRISPR-deactivated Cas9 (dCas9) linked with DNA-methyltransferase 3A's catalytic domain (DNMT3A). When the technique was applied to dopaminergic neurons produced from human induced pluripotent stem cells (hiPSCs) from a PD patient with the SNCA triplication, SNCA mRNA and protein were finely downregulated through targeted DNA methylation in intron 1 [51]. The SNCA triplication hiPSC-derived dopaminergic neurons' disease-related cellular phenotypic characteristics, such as mitochondrial ROS (reactive oxygen species) generation and cellular survival, were also restored by the guide RNA (gRNA)-dCas9-DMNT3A system's reduction in SNCA levels. They discovered that DNA hypermethylation at SNCA intron 1 allows for a viable and adequate tight downregulation of SNCA expression levels, implying that this target sequence joined with CRISPR-dCas9 technology has the potential for precise epigenetic-based remedial methodology for Parkinson's disease [51].
5.3 Huntington’s Disease
Huntington's disease (HD), the most common hereditary neurodegenerative condition is characterised by irrational, excessive motor motions as well as cognitive and emotional problems. The huntingtin (Htt) protein experiences an immoderately long polyglutamine (polyQ) expansion due to the mutation of the HTT gene which is found on chromosome number 4, which gives mutant Htt (mHTT) one or more toxic activities that cause neurodegeneration [52].
One major therapeutic approach to HD is the reduction of mutant HTT mRNA. Taking this into account Morelli and team designed a mutant allele-sensitive CAGEX RNA-targeting CRISPR–Cas13d system (Cas13d–CAGEX). This approach removed toxic CAGEX RNA from fibroblasts produced from HD patients and induces neurons derived from pluripotent stem cells. The study demonstrated that in the striatum of heterozygous zQ175 mice, a model of HD, intrastriatal administration of Cas13d–CAGEX via an adeno-associated viral vector preferentially decreases mutant HTT mRNA and protein levels. Additionally, this resulted in less mutant HTT protein aggregation, less striatal atrophy, and better motor coordination. These phenotypic enhancements persisted with negligible off-target transcriptome impacts and without side effects for a minimum of eight months. These results combined show that an RNA-targeting CRISPR–Cas13d system can be used as a therapeutic strategy for HD [53].
Following in vivo delivery of the mutant HTT gene to the striatum in the R6/2 mouse model of HD, the Cas9 nuclease from Staphylococcus aureus, a small Cas9 ortholog that can be packaged with a single guide RNA into a single adeno-associated virus (AAV) vector, can be used to disrupt the expression of the mutant HTT gene [54]. They discovered that the mutant HTT gene could be disrupted using CRISPR-Cas9, which reduced neuronal inclusions by around 50% and greatly increased longevity and certain motor impairments. [54].
Adult HD140Q-knockin mice's reduced mHTT expression in striatal neuronal cells did not impair viability but did lessen motor impairments. Therefore, the studies reveal that polyglutamine expansion-mediated neuronal toxicity in the adult brain may be effectively and irreversibly removed by non-allele-specific CRISPR/Cas9-mediated gene editing [55].
Shin et al., in 2016, demonstrated the effectiveness of their innovative approach to inhibit the mutant allele in Huntington's disease (HD), caused by a harmful dominant gain-of-function CAG expansion mutation. In order to increase allele specificity, they combined an in-depth understanding of the huntingtin gene haplotype structure with a novel personalized allele-specific CRISPR/Cas9 approach based on PAM-altering SNPs to target patient-specific CRISPR/Cas9 sites with the goal of inactivating only the mutant HTT allele for a specific diplotype [56]. They then, as a “proof-of-concept”, simultaneously removed 44 kb of DNA from the promoter region, transcription start site, and CAG expansion mutation of the mutant HTT gene using two CRISPR/Cas9 guide RNAs (gRNAs) that rely on PAM sites created by SNP alleles on the mutant chromosome. This deletion on the disease chromosome entirely stopped the production of mutant HTT mRNA and protein, clearly showing that the HD mutant allele has been permanently inactivated [56].
5.4 Amyotrophic Lateral Sclerosis
A neurodegenerative condition called amyotrophic lateral sclerosis (ALS) is brought on by the deterioration of motor neurons in the brain and spinal cord [57][58]. There are numerous genetic causes for ALS; at least 20 genes have been linked to the disease. Variants in the SOD1, C9orf72, FUS, and TARDBP genes account for the majority of familial and sporadic instances of ALS. CRISPR/Cas9 genome editing can shed light on the genetics and pathophysiology of ALS. CRISPR/Cas9 has been utilized to validate the impact of ALS-associated mutations and examine phenotypic contrasts between gene-corrected and patient-derived induced pluripotent stem cells (iPSCs). These corrections were made in animal models and patient-derived induced pluripotent stem cells (iPSCs) [58].
Piao and co-workers designed a dual-gRNA design outside of the low complexity region that allowed them to cut the repeat DNA out using a 50% fusion efficiency by the "cutting-deletion-fusion” method. While having minimal impact on C9ORF72 expression this dual-gRNA strategy reduced off-target effects. Their method eliminated the repeat DNA and corrected the RNA foci in neurons containing the patient's C9ORF72 expansion both in vitro and in vivo. As a result, this ‘proof-of-concept’ design corrects the C9ORF72 repeat expansion and may be useful for patients as a potential therapeutic tool [59].
Deng et al., (2021) demonstrated that in two different SOD1-ALS transgenic mice models, CRISPR/Cas9-mediated genome editing reduced the onset of ALS-like pathology and disease. Data provided preclinical proof of the therapeutic method using CRISPR/ Cas9's effectiveness and long-term safety [60].
5.5 Autism Spectrum Disorder
Autism is a spectrum of clinically and etiologically varied diseases that can only be identified by its complicated behavioral manifestation [61]. Identification of relevant genes is challenging due to the large range of variation in ASD (autism spectrum disorder) genotypes and manifestations. There may be hundreds of genes that are very susceptible to de novo mutations, which, while not directly linked to the underlying etiology, may enhance the likelihood of ASD incidence [61]. A number of ASD-related genes are being discovered as a result of next-generation sequencing. Processes such as neuronal activity modulation; transcription and translation regulation; synaptic plasticity; and numerous signaling pathways of biological system are inculpated by these ASD risk genes leading to exhibition of ASD phenotypes [62].
Majority of risk genes for autism spectrum disorder (ASD) are linked to ASD because of haploinsufficiency, a condition in which only one functional copy of a gene exists. In order to counteract the effects of haploinsufficiency, Chen et al., (2024) traced enhancers of two high-confidence autism genes, SCN2A and CHD8, and used CRISPR-based gene activation (CRISPR-A) in hPSC-derived excitatory neurons and cerebral forebrain organoids. In treated neurons and organoids, we discovered that CRISPR-A generated a long-lasting upregulation of CHD8 and SCN2A expression, rescuing gene expression levels and mutation-associated characteristics, such as physiology and gene expression. Hence, supporting a therapeutic intervention of ASD [63].
Tamura and colleagues demonstrated that CRISPR activation (CRISPRa) can offer a promising therapeutic approach by boosting the expression of the existing functional SCN2A allele in the context of SCN2A haploinsufficiency, a significant risk factor for ASD. By demonstrating that restoring Scn2a expression in adolescent heterozygous Scn2a conditional knock-in mice reverses electrophysiological impairments brought on by Scn2a haploinsufficiency, they illustrated the potential for therapeutic benefit. Further, the research team corrected electrophysiological abnormalities in Scn2a heterozygous mice and human stem cell-derived neurons using a therapy based on rAAV-CRISPRa. Their findings concluded that redeeming Scn2a haploinsufficiency can improve neurodevelopmental characteristics, even at teenage stages, and offer a unique therapeutic strategy for many ASD-related genes [64].
RTT (Rett syndrome) patients struggle with severe physical and mental impairments. Methyl-CpG binding protein 2 (MECP2), an X-linked gene encoding an essential epigenetic component for typical nerve cell activity, is heterozygously altered in most RTT patients. RTT syndrome has no known cure, and our knowledge of cellular pathways is still lacking. An effort to create a CRISPR/Cas9-based system that targets and modifies the MECP2 exon 4 coding sequence's disease-relevant regions was made by Le et al., (2019). In human cell lines and iPSCs, they were able to achieve homologous recombination (HR) efficiencies of 20% to 30%. Additionally, in human induced pluripotent stem cells, they successfully introduced a MECP2R270X mutation into the MECP2 gene and were able to effectively correct these mutations in human mutant iPSCs by employing CRISPR/Cas9. In conclusion, they presented a novel approach for MECP2 gene targeting that may be applied to RTT syndrome gene therapy or disease modeling using iPSCs [65].
Rett syndrome is a progressive neurodevelopmental condition that primarily affects females [66]. To fix a hotspot missense mutation in MECP2, c.473 C > T (p.(Thr158Met)), a group of research scientists developed a gene editing toolkit made up of a two-plasmid system. The first construct includes a fluorescent reporter system as well as variant-specific sgRNA and Donor DNA. In order to prevent prolonged Cas9 expression, the second construct adds Cas9 and targets for auto-cleaving. With up to 80% of HDR and fewer than 1% of indels in all cases, NGS analysis on sorted cells from four separate individuals showed an extraordinarily high editing efficiency, indicating the approach's useful potential for treating Rett syndrome [66].
A mutation or deletion of the maternally transmitted UBE3A allele results in Angelman syndrome (AS), a severe neurodevelopmental (a type of ASD) disease. A long non-coding RNA known as UBE3A-ATS silences the patrilineal UBE3A allele in neurons [94]. As part of a thorough screen, Wolter and co-workers discovered that Cas9 may be utilized to target Snord115 genes, short nucleolar RNAs that are grouped in the 3′ region of Ube3a-ATS, to activate paternal Ube3a in cultured mouse and human neurons. During the embryonic and early postnatal periods, when the therapeutic impact of restoring Ube3a is anticipated to be highest, a short Cas9 variant and guide RNA that target around 75 Snord115 genes were supplied to a mouse model of AS. Early intervention restored morphological and behavioral characteristics in AS mice and unlocked paternal Ube3a for at least 17 months in the whole brain [67].
Table 1: CRISPR/Cas-mediated therapeutic approach in treatment of various CNS disorders
|
Target Disease |
Model |
Gene Edited |
Inference |
Reference
|
|
Alzheimer’s Disease (AD)
|
APP/PS1 Mice
|
CysLT1R
|
Improved cognition and conserved synaptic structure and plasticity. Several benefits like reduced inflammation and also supressed kynurenine pathway resulted from CysLT1R deficiency.
|
[40]
|
|
|
Plcγ2-P522R knock-in mouse
|
PLCG2
|
Reduced risk of AD in myeloid lineage cells expressing a rare coding variant in PLCG2 gene.
|
[41]
|
|
|
5XFAD, APP knock-in mouse
|
Bace1
|
70% (approx.) reduction in Bace1 expression in the CA3 region of the treated animals compared to the untreated mice. A significant reduction in β-cleavage products of amyloid precursor protein (APP) in treated 5XFAD brains was also observed.
|
[42]
|
|
|
APP knock-in mice
|
3’-UTR deletion of APP gene
|
Reduced Aβ pathology.
|
[43]
|
|
|
C57BI/6J Mice
|
Mapt (intron -1/exon 1)
|
The young tau knock-out mice had normal memory formation and susceptibility to excitotoxic seizures was also reduced.
|
[44]
|
|
Parkinson’s Disease (PD)
|
6-OHDA-lesioned rats |
th gene |
DA-producing astrocytes can be produced by SAM-induced expression of the astrocyte's endogenous th gene, which effectively lessens the motor asymmetry brought on by the lesion. |
[47] |
|
|
Human iPSC- derived neuron |
SNCA |
CRISPRi downregulation of alpha-synuclein demonstrated functional recovery through decrease of mitochondrial DNA damage and oxidative stress. |
[48] |
|
|
Rat
|
A53T-SNCA
|
The upregulation of α-synuclein, reactive microgliosis, dopaminergic neurodegeneration, and parkinsonian motor symptoms were all dramatically reversed by gene deletion of A53T-SNCA.
|
[49]
|
|
|
Human embryonic stem cell (hESC) line
|
SNCA
|
Reduction or elimination of SNCA alleles, conferring a degree of resistance to Lewy disease.
|
[50]
|
|
|
Human induced pluripotent stem cell (hiPSC) derived dopaminergic neurons
|
SNCA
|
Downregulation of SNCA expression levels by hypermethylation of DNA at SNCA intron 1. The SNCA triplication hiPSC-derived dopaminergic neurons' disease-related cellular phenotypic characteristics, such as mitochondrial ROS generation and cellular survival, were also rescued by reduction in SNCA levels.
|
[51]
|
|
Huntington’s Disease (HD)
|
Heterozygous zQ175 mice |
HTT |
Intrastriatal administration of Cas13d–CAGEX via an adeno-associated viral vector preferentially decreased mutant HTT mRNA and protein levels and also reduced mutant HTT protein aggregation, less striatal atrophy, and better motor coordination.
|
[53] |
|
|
R6/2 mouse |
HTT
|
Mutant HTT gene disrupted using CRISPR-Cas9, reduced neuronal inclusions by around 50% and greatly increased longevity and certain motor impairments.
|
[54]
|
|
|
HD140Q-knockin mice
|
HTT
|
By permanently reducing endogenous mHTT expression in the striatum of mHTT -expressing animals, HTT aggregates were successfully depleted.
|
[55]
|
|
|
Primary fibroblast cells derived from a male Caucasian HD patient
|
HTT
|
Complete inactivation of mutant allele entirely stopped the production of mutant HTT mRNA and protein.
|
[56]
|
|
Amyotrophic Lateral Sclerosis (ALS)
|
HEK293 cells
|
C9ORF72 |
Both in vitro and in vivo elimination of repeat DNA and correction of RNA foci.
|
[59]
|
|
|
SOD1-ALS transgenic mice
|
SOD1
|
Prevention of onset of ALS-like pathology and disease.
|
[60]
|
|
Autism Spectrum Disorder (ASD)
|
hPSC-derived excitatory neurons and cerebral forebrain organoids |
SCN2A and CHD8 |
Long-lasting upregulation of CHD8 and SCN2A expression, rescue of gene expression levels and mutation-associated characteristics.
|
[63] |
|
|
Scn2a heterozygous mice, human stem-cell-derived neurons
|
SCN2A
|
Even at adolescent phases, Scn2a haploinsufficiency rescue can improve neurodevelopmental characteristics.
|
[64]
|
|
|
Mice
|
UBE3A (Snord115) |
Early intervention restored morphological and behavioral characteristics in Angelman syndromic mice and unlocked paternal Ube3a for at least 17 months in the whole brain. |
[67]
|
6. Challenges And Future Prospects
The area of gene therapy is fast growing, and it appears that researchers have only begun to explore its possibilities [68]. Despite CRISPR/enormous Cas's promise for genome editing, a number of critical challenges still need to be resolved, including off-target mutations, PAM dependency, gRNA synthesis, and CRISPR/Cas9 delivery techniques, of which off-target mutations are highly worrisome. Genomic toxicity, cancer, genome instability, disruption of gene function, and epigenetic changes are all results of it [25]. The transfer of therapeutic medicines into the central nervous system is a significant obstacle to the application of in vivo genome editing (CNS) [69]. The CRISPR systems' delivery techniques could possibly affect brain cells and trigger immunological reactions. CRISPR constructs are frequently delivered across the BBB using viral vectors. Viral delivery, on the other hand, results in prolonged CRISPR expression, which harms neuronal cells and changes their morphologies [70]. Notably, less invasive delivery methods that can target bigger brain areas will often be more advantageous for non-human primate research and clinical human trials [5]. PAM is necessary for the CRISPR-Cas9-sgRNA complex to land at the protospacer DNA and begin the target DNA interrogation, which is important for the accuracy of the CRISPR-Cas9 DSB activity. The application of CRISPR-Cas9 technology could sometimes be limited since none of the PAMs discovered so far, or even a combination of all known PAMs could feasibly cover any complete genome sequences [37].
7. CONCLUSION
CRISPR/Cas technology has emerged as a promising tool and has elevated the field of genome editing to new heights. The research field has seen both the benefits and drawbacks of using CRISPR/Cas as a therapeutic tool for challenging medical conditions like neurological diseases. Before using CRISPR/Cas methods to treat human diseases, there are undoubtedly technical challenges and safety concerns that need to be resolved. Nevertheless, preclinical research addressed the conceptual underpinnings and essential elements required for the application of CRISPR/Cas to neurological diseases, and the potential of integrative approaches was also shown. Although it is understood that a number of factors need to be addressed before CNS genome-editing therapies can be established, the area is developing at an astounding rate, bringing potential clinical applications closer day by day.
Ethical approval and consent to participate
Not applicable.
Consent for Publication
Not applicable
Availability of Data and Materials
Not applicable
Competing interests
The authors declare that they have no competing interests.
Funding
This Review Research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors’ Contributions:
The data was collected and written by Dr. Madhurima Tiwari and Shifa Ahmad. The entire article was guided by Dr Deepika delsa Dean. Diagrams and proofreading of the manuscript done by Ms. Poonam.
Conflict of interest
The authors declare no conflict of interest
REFERENCES




Madhurima Tiwari, Shifa Ahmad, Poonam Tripathi, Deepika Delsa Dean*, CRISPR/CAS Technology: A Promising Tool For The Treatment Of Central Nervous System Disorders, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 3656-3677. https://doi.org/10.5281/zenodo.21432759
 10.5281/zenodo.21432759
10.5281/zenodo.21432759