
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Sciences, RIMSR, Puthuppally, Kottayam, Kerala, India 686009.
Delivering therapeutics across restrictive biological barriers remains a primary challenge in pharmaceutical development. Conventional nanocarriers like liposomes and niosomes suffer from chemical instability and rigid membranes that restrict the drug to be delivered into deep tissues. Bilosomes resolve these issues by integrating bile salts as structural edge activators into a non-ionic surfactant or phospholipid matrix stabilized by cholesterol, these advanced nanovesicles maintain their integrity against enzymatic degradation and shifting pH gradients. Simultaneously, they have stress-responsive elasticity, enabling the intact vesicles to reversibly deform and traverse tight intercellular junctions smaller than their own diameter. This review comprehensively examines different bilosomal formulation strategies, scalable preparation techniques and solid-state probilosome transitions for extended shelf-life. It also explores advanced surface modifications, including PEGylation and active ligands for targeted delivery. Finally, this review highlights the multi-route versatility of bilosomes, demonstrating their capacity to enhance the bioavailability, pharmacokinetic profiling, and targeted efficacy of diverse therapeutics across oral, transdermal, ocular, intranasal, and parenteral administration. The engineered bilosomes would help it from being a supramolecular design and making it clinically applicable, offering a highly adaptable platform for next-generation, route-specific drug delivery.
Drug delivery across biological barriers remains a fundamental challenge in pharmaceutical development. Oral administration exposes drugs to harsh gastric pH, enzymatic degradation, and the intestinal mucus layer before absorption[1]. Systemic drugs that survive gastrointestinal transit encounter additional obstacles: tight cellular junctions, first-pass hepatic metabolism, and, for some molecules, renal clearance. Topical and ocular routes present their own barriers like the stratum corneum and tear-fluid washout, respectively[2]. These barriers particularly limit delivery of poorly soluble small molecules (BCS Class II and IV) and fragile therapeutics such as peptides and nucleic acids[1].
Researchers have long encapsulated drugs within nanocarrier systems to shield molecules from degradation and alter systemic uptake and tissue distribution. However, rigid nanocarriers cannot penetrate restrictive tissues, which demands that carriers possess ability to reversibly deform and travel pores smaller than their own diameter[3].
Limitations of Conventional Vesicular Carriers
Liposomes were the first vesicular carriers widely adopted for drug delivery. Despite their biocompatibility, liposomes suffer from poor chemical stability. Natural lipids oxidize during storage, and upon oral administration, bile salts in the intestinal lumen destabilize the bilayer, leading to premature drug release[4]. To address oxidation, niosomes were developed by replacing phospholipids with synthetic, non-ionic surfactants. That modification imparts stability against long-term storage and gains chemical stability[5]. However, niosomes are far more rigid than liposomes. The stiff bilayer membrane cannot deform, preventing penetration through restrictive barriers such as intestinal tight junctions or dermal pores. This loss of mechanical flexibility severely limits niosomal delivery to shallow tissue layers[6].
Overcoming Delivery Barriers
Bilosomes resolves the trade-off between stability and deformability. They combine synthetic surfactants with bile salts (e.g., sodium deoxycholate or sodium taurocholate) and function as structural edge activators. Bilosomes exhibit the elasticity required for vesicles to deform and travel narrow biological pores. This includes intestinal tight junctions, dermal lamellae, and subcellular compartments all while retaining their integrity and encapsulated payload. The combination of chemical stability with membrane deformability is the defining feature of bilosomes[6].
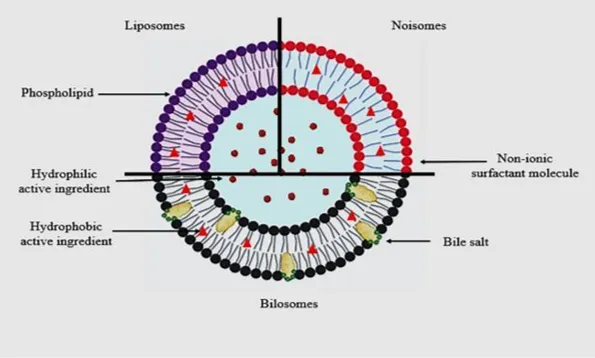
Fig.No.1. Diagrammatic depiction of nanovesicular drug delivery systems, specifically liposomes, niosomes, and bilosomes[6].
Composition of Bilosomes
Surfactants and Phospholipids
Non-ionic surfactants provide the core structure for bilosomes. They have low toxicity and good biocompatibility. Span 60 is the standard choice for formulation as its fully saturated hydrocarbon chain has a high phase transition temperature, keeping the structure stable and payload intact[7]. Span 40, Span 20, and Span 80 are also used as they can adjust membrane fluidity for different tissue barriers[2]. Polymeric surfactants such as Poloxamer 188 increase entrapment efficiency by establishing a hydrogen-bonding network within the vesicular core.
Natural phospholipids like purified lecithin or soybean phosphatidylcholine can eliminate the need for synthetic surfactants[8]. They can form bilayers that can mimic native cell membranes, which improves biocompatibility and protects the payload against enzymatic degradation. Both approaches require the incorporation of bile salts to establish the membrane fluidity necessary for lipophilic compound delivery[4].
Bile Salts as Edge Activators
Bile salts are integrated into the lipid bilayer as both structural fluidizers and edge activators. Their rigid steroid core disrupts surfactant tail packing and lowers the phase transition temperature, converting the membrane to a fluid, liquid-crystalline state[9]. Edge activators accumulate at high-curvature regions, reduce surface tension, and enable vesicles to deform and pass through pores one-tenth of their diameter without rupture. Bile salts also stabilize the vesicles against acidic pH and enzymatic attack[10].
The choice of bile salt determines the carrier’s function. Sodium deoxycholate (SDC) enhances permeation for topical and oral delivery[11]. For oral use, conjugated bile salts are superior. Sodium taurocholate and sodium glycocholate improve gastrointestinal retention, with sodium glycocholate lowering toxicity[12]. Ursodeoxycholic acid (UDCA) provides membrane flexibility and intrinsic anti-inflammatory activity[13]. The negative charge imparted by bile salts also provides colloidal stability and prevents vesicle aggregation[6].
Cholesterol and Bilayer Stabilization
Excessive membrane fluidity from edge activators can destabilize the vesicle, promoting fusion or early payload release. Cholesterol is added to counteract this[14]. It inserts between non-ionic surfactant molecules, restricting tail mobility and raising the phase transition temperature. The result is a stiffer membrane with lower permeability and fewer aqueous pores, which reduces payload leakage, especially for hydrophilic compounds[1].
Cholesterol concentration has a greater effect on both entrapment entrapment efficiency (EE%) and vesicular stability. Higher cholesterol levels improve sustained release by slowing drug diffusion and protects against enzymatic degradation and acidic pH. Cholesterol and bile salts show opposing effects on the lipid bilayer, creating a functional balance: cholesterol maintains structural rigidity, while bile salts provide the required membrane fluidity[10]. Optimal molar ratios of surfactant, bile salt, and cholesterol maintain the 100–300 nm vesicle size critical for tissue penetration and payload retention[1].
Preparation Methods
Formulation methods are categorized into conventional solvent-based methods, scalable high-energy techniques, and solid-state engineering strategies. The optimal technique is selected based on the physicochemical properties of the drug and the required stability of the final dosage form.
Solvent-Based Methods
Thin-Film Hydration (TFH)
Thin-Film Hydration (TFH) involves dissolving non-ionic surfactants, cholesterol, and the drug in a volatile organic solvent like chloroform. The solvent is removed via rotary evaporation under reduced pressure at 40–60°C, depositing a homogenous lipid film on the flask wall. The film is then rehydrated using an aqueous bile-salt buffer under gentle stirring for 30-60 minutes. This hydration step must occur above the surfactant’s gel-to-liquid phase transition temperature (Tc ) to induce membrane curvature and form multi-lamellar vesicles (MLVs). Finally, probe sonication is used to reduce the MLVs to the optimal 100–300 nm size range[7].
Reverse-Phase Evaporation
This technique produces large unilamellar vesicles (LUVs) to improve the entrapment of hydrophilic compounds. Surfactants and cholesterol are dissolved in an organic solvent, followed by the addition of an aqueous phase containing the drug and bile salts. Sonication of this biphasic system produces a stable water-in-oil (w/o) emulsion. Subsequent solvent removal under vacuum causes the emulsified droplets to collapse into a viscous, gel-like thin lipid film, which must then be hydrated with an aqueous phase to form the final uniform colloidal dispersion of LUVs[1].
Solvent Evaporation
The payload, surfactant, cholesterol, and bile salts are co-dissolved in a volatile mixed solvent system, typically methanol and methylene chloride. Instead of applying a vacuum, the solution undergoes continuous magnetic stirring at an elevated temperature (e.g., 60°C) until the solvents completely evaporate. The resulting dry residue is directly reconstituted with an aqueous buffer, driving spontaneous self-assembly into a bilosomal suspension[4].
Ethanol Injection
This rapid technique relies on solvent displacement and nanoprecipitation. The payload, surfactants, and cholesterol are dissolved in ethanol. This organic phase is rapidly injected into a pre-heated (50–60°C), magnetically stirred aqueous phosphate buffer (pH 7.4) containing dissolved bile salts. Immediate turbidity signals spontaneous vesicle formation. The dispersion is stirred for several hours to eliminate residual ethanol via evaporation or dialysis, followed by cooling and probe sonication for size refinement[4].
Solvent-Free and High-Energy Techniques
Hot Homogenization (Melt Method)
This solvent-free technique is well suited for encapsulating lipophilic drugs. Lipid components (e.g., 1-monopalmitoyl glycerol, cholesterol, and dicetyl phosphate) are heated to 130°C-140°C to form a uniform melt. The lipophilic payload dissolves directly into this molten phase. A pre-heated aqueous bile-salt buffer is then added, and high-pressure homogenization induces vesiculation. For heat-labile macromolecules like protein antigens, the payload is introduced after homogenization. Encapsulation is then achieved through multiple freeze-thaw cycles to prevent thermal degradation[4].
Microfluidization
Representing a scalable, industrial-level technology, microfluidization utilizes precisely controlled fluid dynamics to generate nanovesicles continuously. An organic phase (lipids, surfactants, and bile salts dissolved in a water-miscible solvent) and an aqueous phase are pumped through specialized microchannels at strictly controlled flow rates and extreme pressures. At the microchannel junction, rapid mixing, high shear rates, and sudden solvent exchange causes the components to instantaneously precipitate and self-assemble into highly uniform bilosomes. This method is highly valued because it bypasses the need for post-processing size reduction and yields a very narrow particle size distribution[8].
Microwave-Assisted Synthesis
Microwave-assisted synthesis of bilosomal vesicles eliminates the need of organic solvents from its preparation. It utilizes controlled microwave irradiation to rapidly deliver the precise thermal energy required to drive lipid self-assembly. The method demonstrates advantages for temperature-sensitive formulations such as vaccines, where synthesis time and cargo integrity are critical constraints[6].
Solid-State and Pro-Vesicular Engineering
Lyophilization
To prevent long-term instability such as drug leakage, vesicular aggregation, and chemical degradation, liquid bilosomes are converted into dry probilosome powders[15]. The dispersion is first mixed with a cryoprotectant, such as trehalose or spray-dried mannitol, to prevent ice-crystal-induced membrane rupture [9,16]. The mixture should be pre-frozen at -20°C for 24 hours and then subjected to extreme vacuum lyophilization (e.g. 0.035 mbar at -60°C). These stable probilosomes extends shelf-life and facilitates formulation into solid oral dosages. Upon contact with aqueous media, the powder rehydrates directly into intact bilosomes[15].
Slurry Method
The vesicle-forming components (lipids, bile salts, and the drug payload) are co-dissolved in an organic solvent. This organic mixture is poured directly over a highly water-soluble carrier powder, such as sorbitol, mannitol, or maltodextrin. The solvent is then completely removed via vacuum drying in a rotary evaporator, leaving the carrier particles uniformly coated with a dry lipid film [2].
Post-Formulation Size Reduction
Regardless of the preparation technique used, crude dispersions often result in large, heterogeneous vesicles that must be downsized to enable effective biological permeation.
Sonication: MLV dispersions are subjected to probe sonication or ultrasonic water-bath sonication to shatter large vesicles into smaller, uniform unilamellar vesicles[5,17].
Membrane Extrusion: The dispersion is forced through polycarbonate filters with strictly defined pore sizes (e.g., 100 nm or 200 nm), standardizing the vesicle size without generating the extreme localized heat associated with probe sonication[18].
High-Pressure Homogenization: Uses mechanical shear to ensure uniform size distribution and modify lamellarity, proving highly scalable and beneficial for preserving sensitive peptides without relying on organic solvents[6].
Characterization and Evaluation
Vesicle Size and Polydispersity Index
Dynamic Light Scattering (DLS) determines hydrodynamic diameter and size distribution. To penetrate restrictive barriers such as the stratum corneum or intestinal M-cells, bilosomes must maintain diameters between 100 and 300 nm, ideally under 200 nm[19]. Specific formulation components control these dimensions, high-transition-temperature surfactants (e.g., Span 60) reduce vesicle size, while excess cholesterol or bile salts increase it[20]. The Polydispersity Index (PDI) measures size uniformity. A PDI ≤ 0.3 confirms a stable, monodisperse system, whereas values exceeding 0.5 indicate physical instability and vesicle aggregation[21].
Zeta Potential and Colloidal Stability
Zeta Potential (ζ ) is a primary indicator of colloidal stability. Anionic bile salts (e.g., sodium deoxycholate) causes the lipid bilayer to have a strong electronegative charge[7]. Formulations with zeta values exceeding |±30| mV resist flocculation through electrostatic repulsion. This charge induction also fluidizes the lipid matrix, providing the plasticity and deformability required for the vesicle to pass through tight biological junctions[9,20].
Morphological Imaging (TEM, SEM, AFM)
Transmission Electron Microscopy (TEM) with negative staining (e.g., 1% w/v phosphotungstic acid) confirms the presence of closed spherical bilayers and provides physical size measurements[22]. Scanning Electron Microscopy (SEM) details surface topography, verifying complete encapsulation by the absence of unentrapped drug crystals on the exterior[7]. Atomic Force Microscopy (AFM) maps 3D topology without chemical stains and measures the elasticity and deformability imparted by bile salt edge activators[23].
Entrapment Efficiency and Drug Loading
Cooling ultracentrifugation (e.g., 20,000 rpm for 90 minutes) separates unentrapped drug from the loaded bilosomes[14]. High-Performance Liquid Chromatography or UV-Visible spectrophotometry quantifies the free drug in the supernatant. Entrapment Efficiency is calculated as:
EE%=Total Drug Added-Free Unentrapped DrugTotal Drug Added×100
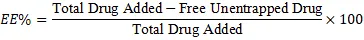
Integrity of lipid matrix dictates retention. Surfactants with high phase transition temperatures (e.g., Span 60) and moderate cholesterol levels increase bilayer rigidity and payload retention. In contrast, excess cholesterol causes steric hindrance, and excess bile salts disrupt the lamellar architecture. Both conditions lead to payload leakage and reduce overall entrapment efficiency[15].
In Vitro Release Studies
Oral formulations utilize the dialysis membrane bag method using cellulose barrier, whereas topical and ocular formulations rely on Franz diffusion cells separated by synthetic membranes. Sink conditions are maintained across all setups by replacing withdrawn aliquots with equivalent volumes of fresh medium[24]. The drug concentration is quantified via High-Performance Liquid Chromatography (HPLC) or UV-Visible spectrophotometry.
Oral administration is simulated at 37°C for 2 hours in Simulated Gastric Fluid (pH 1.2), followed by 4-6 hours in Simulated Intestinal Fluid (pH 6.8)[25]. For topical and ocular applications, the Franz cell receptor compartment utilizes phosphate-buffered saline (pH 7.4), maintained at 32°C for dermal conditions or 37°C for mucosal and corneal targets[17,26].
Ex Vivo Permeation Studies
Oral absorption and permeability are assessed through the non-everted rat intestinal sac or in situ single-pass perfusion models. For transdermal, buccal, and ocular routes, excised tissues (e.g., Wistar rat skin, porcine mucosa, or rabbit corneas) are mounted in Franz diffusion cells.
Analysing the receptor aliquots yields steady-state flux (Jss), permeability coefficient (Kp), and the enhancement ratio (ER)[27]. A Deformability Index quantifies the vesicle's capacity to traverse mucosal or stratum corneum barriers intact[28]. Post-permeation histological evaluation of excised tissues verifies bile salt-mediated tight junction disruption for paracellular transport without cytotoxic structural cell loss[29].
Advanced Bilosomal Engineering
To tailor bilosomes for specific routes and overcome complex biological barriers, their basic structure can be modified extensively. These strategies range from surface shielding with polymers to the integration of active ligands for targeted cellular uptake.
Polymeric and Polysaccharide Coatings
Chitosan-Coated and Core-Shell Nanoparticles
Chitosan is a natural, polycationic biopolymer that can serve either as a simple surface coating or as the core of a more complex hybrid structure. For surface coatings, chitosan is deposited onto negatively charged bilosome through electrostatic adsorption. In bilosome-chitosan nanoparticle structures, positively charged chitosan core is first formed by ionic gelation. When mixed, the electrostatic interaction induces the bilosomal membrane to reorganize around the chitosan core, fully encapsulating it. Trehalose (5%) is added as a cryoprotectant to preserve this core-shell morphology during freeze-drying.
The mechanism of mucosal penetration depends on charge. Chitosan's cationic surface interacts with negatively charged mucin glycoproteins, temporarily opening tight epithelial junctions to allow paracellular transport. The resulting core-shell structure can carry both hydrophilic drugs (e.g., daclatasvir) and hydrophobic ones (e.g., xanthone) simultaneously, effectively overcoming the rapid clearance of traditional vesicles[16].
Hyaluronic Acid for Mucoadhesion
Hyaluobilosomes are formulated by dissolving hyaluronic acid (HA), a bioadhesive amphiphilic polymer, into the preheated aqueous bile salt solution (0.25% w/v) before the hydration step. For ocular delivery, HA binds to corneal mucin, increasing formulation viscosity and prolonging residence time at the ocular surface. This reduces the tear-drainage clearance of drugs like felodipine and agomelatine used in glaucoma management[28]. Beyond ocular applications hyaluobilosomes are used to target inflamed areas for the treatment of arthritis[30].
Dextran Modifications for Stealth and Macrophage Uptake
Dextran sulfate is a highly anionic, hydrophilic polysaccharide. It is coated onto positively charged bilosomes (surface charge introduced via stearylamine) through a titration process utilising strong electrostatic interactions. The resulting dense polysaccharide layer creates a steric barrier against opsonization, reducing immune recognition. Beyond passive stealth, DS actively targets scavenger receptor type A (SR-A) on macrophages within atherosclerotic plaques, triggering receptor-mediated endocytosis while competitively inhibiting the uptake of modified LDL[13].
A different approach utilizes cationic dextran derivatives. Diethylaminoethyl dextran is used to coat bilosomes to synchronize the biodistribution of co-delivered drugs such as berberine and curcumin in non-alcoholic fatty liver disease models, ensuring the two agents reach the same tissue compartments at therapeutically relevant ratios.[9].
PEGylated Bilosomes for Opening Buccal Tight Junctions
These PEGylated bilosomes are designed to modulate intercellular junctions through the integration of PEG2000 and Sodium Glycodeoxycholate (GDC). When applied to the buccal mucosa, QCM-D analysis demonstrates they form reversible interactions with mucin and stable interactions with phosphatidylcholine on epithelial cell surfaces. To facilitate drug delivery, GDC enhances membrane fluidity to temporarily disrupt cell junctions. It also chelates calcium ions, reversibly weakening calcium-dependent desmosomes. Together, these mechanisms allow the drug to transit through both paracellular and transcellular pathways without inducing permanent epithelial tissue damage[29].
pH-Responsive Layer-by-Layer Coating with Polygalacturonic Acid
Polygalacturonic acid is a pH-responsive anionic biopolymer used in layer-by-layer assembly for functional food applications. To form the double coat, chitosan-coated bilosomes are added dropwise into a PGA solution under stirring, forming a second outer layer over the existing chitosan shell.
This pH sensitivity represents the core mechanism of the formulation. In acidic gastric fluid, PGA carboxyl groups protonate to form a compact, hydrogen-bonded gel that seals the payload and protects it within complex food matrices (e.g., oat milk), masking the bitter taste of bile salts in the process. In the alkaline intestine, those same groups deprotonate and generate electrostatic repulsion that swells the shell and drives the drug to be released at the target site. The same structural change that protects in the stomach triggers release in the gut[31].
Pluronic P123 and TPGS as Efflux Pump Inhibitors
These modifications involve the incorporation of a triblock copolymer (PEO-PPO-PEO) such as Pluronic P123, or D-alpha-tocopheryl polyethylene glycol succinate (TPGS). These are co-assembled alongside phosphatidylcholine, cholesterol, and bile salts to create a stealth surface shell around the bilosome.
Pluronic P123 introduces specific thermal sensitivity to the bilosome. When exposed to the elevated heat and acidity of a tumor microenvironment, the PEO blocks of the polymer undergo hydrolytic degradation. The elevated temperature causes the phospholipid bilayer to shift from a gel phase to a liquid phase[32]. TPGS works through a different mechanism by inhibiting P-glycoprotein, the efflux pump that exports chemotherapeutics out of cancer cells[3]. In multidrug-resistant breast cancer models, this inhibition substantially increases the intracellular drug concentration and cytotoxicity[2].
Active Targeting and Protein Integration
Galactosylated Stearylamine for Hepatic Targeting
GSA is synthesized by coupling galactose-containing lactobionic acid to stearylamine via amide bond formation. The resulting molecule is integrated into the lipid bilayer during ethanol injection method, positioning the galactose group on the nanoparticle surface. Galactose is used because it selectively targets asialoglycoprotein receptors (ASGPRs), which have carbohydrate recognition domains found almost exclusively on hepatocytes. This receptor-ligand interaction triggers endocytosis and delivers drugs like sofosbuvir into the liver to treat Hepatitis C[33].
Dextrose Adsorption for Lectin Receptor Targeting
Dextrose (glucose) is coated onto fully formed, drug-loaded bilosomes via polymeric adsorption. It targets lectin-carbohydrate receptors on the plasma membrane of hepatocytes and spleen cells, driving receptor-mediated endocytosis and forcing cellular accumulation in the liver for hepatocellular carcinoma or chemically induced liver injury[9]. The coating also provides strong stability against digestive enzymes in the gastrointestinal tract[34].
Folate-PEG Conjugates to Overcome Multi-Drug Resistance
The FA-PEG-CHS construct utilizes a hydrophilic PEG chain to connect the folic acid targeting ligand directly to the cholesterol anchor. During formulation, the cholesterol anchor embeds in the bilayer while the PEG spacer arm extends outward, projecting the folic acid into the aqueous space. Folic acid binds to folate receptors, which are overexpressed on breast cancer, ovarian, and lung tumor cells. This binding triggers receptor-mediated endocytosis, allowing chemotherapeutics (e.g: vorinostat) to bypass drug efflux pumps and overcome multi-drug resistance[30].
Glucomannan Anchoring for Oral Vaccine Delivery
This functionalized lipid combines a carbohydrate ligand (glucomannan) with a hydrophobic phospholipid (DSPE). Unlike the simple surface coatings, the lipophilic DSPE tail anchors deep in the bilayer, leaving the glucomannan head exposed to aqueous exterior and remains firmly attached[10]. The mannose-targeting ligands enable the bilosome to specifically engage receptors on antigen-presenting cells and M-cells. This allows the safe delivery of oral vaccines across the mucosal barrier and elicit strong IgA and IgG immune responses[4].
Co-Delivery with Bioactive Peptides (Zein, Melittin, Lactoferrin)
Bioactive proteins and cytolytic peptides can be integrated directly into the bilosomal structure. Zein (maize protein) and melittin are embedded in the bilayer, while lactoferrin is anchored to the surface. Zein provides exceptional hydrophobicity, stabilizing the membrane and enhancing cytostatic and apoptotic activity against lung cancer cells[35]. Melittin works synergistically with chemotherapeutics (e.g. icariin), amplifying pro-apoptotic activity against pancreatic cancer cells[2]. Lactoferrin targets receptors on hepatocellular carcinoma cells, driving endocytosis and cellular uptake[36].
Bilayer and Core Modifications
Reversing Surface Charge with Cationic Lipids
Bilosomes naturally carry a negative charge, but this modification reverses the zeta potential by substituting standard lipids with cationic lipids such as stearylamine (SA) or DOTAP. The positive surface charge promotes electrostatic attraction and membrane adhesion to the negatively charged cell membranes and mucosal layers, increasing cellular internalization. To achieve selective hepatic targeting, stearylamine is chemically conjugated with galactose to form galactosylated stearylamine (GSA), which actively targets the anionic carbohydrate recognition domains of ASGPRs on hepatocyte. The high positive charge also creates electrostatic repulsion between vesicles in the stomach, improving acid resistance compared to standard negative bilosomes[33].
Using Hydrogenated and Fat-Free Phospholipids
Replacing standard lipid bases with highly modified phospholipids improves the consistency of the bilosome structure. Phospholipon 80H (hydrogenated soybean phospholipids containing 70% phosphatidylcholine) and Lipoid S45 (fat-free soybean lecithin containing 45% phosphatidylcholine ) serve as the bilayer backbone. Hydrogenation and fat removal eliminate structural defects and oxidation vulnerability found in natural lipids. This supports the production of small, stable vesicles with excellent tissue penetration. This approach is valuable for deep pulmonary delivery in acute lung injury[37].
Incorporating Ultra-Deformable Edge Activators
Edge activators are elastic surfactants consisting of single-chain, pegylated, or polyoxyethylated compounds like Brij C2, Cremophor EL, or essential oil terpenes (limonene, citral) that are co-assembled into the bilayer alongside bile salts. These compounds lower the interfacial tension of the bilayer. Their hydrophilic PEG moieties absorb water from the stratum corneum, causing the skin's lipid structure to swell and widen intracellular barrier junctions. This allows the bilosome to improve its permeability through the tight-junctions which are smaller than their own diameter[27].
Poloxamer 188 Integration for Vesicle Size Reduction
Rather than acting as an external gelling agent, Poloxamer 188 is integrated directly into the vesicular membrane. It is added into the aqueous hydration phase along with bile salts to co-assemble into the bilosome structure. Poloxamer 188 alters the physical bulk of the vesicle. It modulates hydrophilic/lipophilic balance of the system, resulting in an inverse relationship with vesicle size, shrinking the vesicle. Synergistically, the hydrophobic PPO chains within the poloxamer interact with the drug, drastically increasing the entrapment efficiency of lipophilic drugs[38].
Bile Acid Substitutions and Conjugates
Alternative Therapeutic Bile Acids (UDCA) & Lithocholic Acid (LCA)
This modification uses Ursodeoxycholic Acid (UDCA) or Lithocholic Acid (LCA) as the core bile salt instead of conventional sodium deoxycholate. They are added alongside the phospholipids during thin-film hydration. UDCA acts as a biomaterial-tethered carrier with active biological effects, capable of dissolving atherosclerotic cholesterol crystals and causing the regression of developed arterial plaques[13]. LCA possesses anti-cancer properties, exerting cytostatic effects on breast cancer cells, inducing mesenchymal-to-epithelial transition. It enhances the anti-tumor immune response synergistically with drugs like vorinostat[30].
Taurine and Glycine Conjugates for Gastric Resistance
This structure involves the use of bile salts conjugated with taurine or glycine as the primary structural edge activator during hydration. Conjugating bile acids with taurine increases the polarity and acidity of the side chains, lowering their pKa down to ~2. This modification prevents bile salts from precipitating in highly acidic gastric environments, making them superior for maintaining vesicular elasticity and stability across shifting pH gradients[12].
Chylomicron-Mimicking Emulsomes for Lymphatic Uptake
These are bio-engineered, self-assembling carriers mimicking natural chylomicrons, with a solid or semi-solid lipid core surrounded by phospholipid layers integrated with bile salts[39]. Emulsomes are absorbed through the intestinal lymphatic system similar to dietary lipids. This lymphatic transport pathway allows the encapsulated drugs to successfully bypass first-pass liver metabolism, improving their oral bioavailability[12].
In Situ Systems and Polymeric Matrices
Self-Emulsifying Phospholipid Pre-concentrates (SEPPs)
SEPPs are anhydrous, liquid pre-concentrates of bilosomes. Lipids, surfactants, bile salts, and drugs are dissolved in a tailored solvent/co-solvent mixture to form a stable liquid. Upon oral ingestion, the liquid self-assembles into nanometric bilosomes on contact with the aqueous gastrointestinal fluids. This eliminates complex manufacturing steps and solves long-term aggregation issues, making it suitable for large-scale pharmaceutical capsule filling[12].
Formulating Temperature and pH-Triggered In Situ Gels
These are stimuli-responsive polymeric matrices. Liquid bilosomes are dispersed into polymer networks such as Poloxamer 407/188/Carbopol 971P or gellan/xanthan gum. The formulations remain liquid at room temperature but undergo phase transition into a viscous, mucoadhesive gel upon exposure to body temperature, pH shifts, or tear-fluid electrolytes. This alters diffusion path lengths and governs drug release according to non-Fickian transport and matrix erosion, prolonging residence time in the eye or nasal cavity for direct nose-to-brain delivery[10].
Solid Polymeric Ocular Inserts & Mucoadhesive Sponges
Solid inserts and sponges are cast from bilosomal dispersions using solvent evaporation method with film-forming polymers such as HPMC, Sodium Alginate, or Sodium CMC. Placed in the conjunctival cul-de-sac or buccal cavity, these devices circumvent the rapid drainage and tear turnover of liquid drops. They provide controlled, zero-order, extended release of the payload over several hours via polymer swelling and gradual dissolution[11,21].
Theranostics, Magnetic, and Subcellular Delivery
Radiolabeling with Iodine-131 for In Vivo Tracking
This modification transforms the bilosome into an advanced theranostic device. The encapsulated payload, such as resveratrol, is radiolabeled via electrophilic substitution with Iodine-131. This enables real-time gamma-scintigraphy tracking in vivo, providing quantitative data on tumor accumulation, biodistribution, and pharmacokinetic clearance. The imaging directly demonstrates that bilosomes exploit the Enhanced Permeability and Retention (EPR) effect to accumulate in tumor tissues at rates higher than free drugs[40].
Magnetic Guidance using SPIONs and Pluronic Modifiers
Superparamagnetic Iron Oxide Nanoparticles (SPIONs) are encapsulated within the bilosomal core, surface-functionalized with molybdenum disulfide (MoS₂), and decorated with Pluronic F126-MA or F127-MA[41]. When an external magnetic field is applied, these vesicles are directed across biological barriers, such as the olfactory mucosa, for nose-to-brain delivery in Alzheimer's treatments[42]. Pluronic F126/127-MA also targets gastrointestinal cancers by interfering with KRAS mutations, reducing cancer cell motility and invasion[41].
Lipodextrin Conjugates for Deep Tumor Penetration
This modification integrates specific G/lipodextrin conjugates into the bilosomal lipid matrix. Lipodextrin moieties are conjugated into the bilayer alongside chemotherapeutics such as doxorubicin, enabling intracellular penetration of solid tumors. While standard bilosomes enhance general absorption, G/lipodextrin enrichment provides a specific mechanism for accessing the intracellular space, resulting in substantially higher cytotoxicity than the free drug alone[41].
Montmorillonite Clay Nanodisks for Endosomal Escape
These are hybrid inorganic-organic nanocarriers composed of montmorillonite clay nanodisks stabilized by a polycation-chitosan layer and integrated with bile salts such as sodium cholate or deoxycholate. The clay matrix acts as a pH-triggered nanocarrier designed to co-deliver multiple chemotherapeutics, such as fluorouracil and doxorubicin, into the acidic endosomal compartments of tumor cells[41].
Barrier-Focused Engineering of Bilosomal Nanocarriers
|
Barrier & objective |
Engineered modification |
Core mechanism |
Clinical outcome & drugs example |
|
ORAL DELIVERY |
|||
|
Gastrointestinal stability |
Probilosomes (Lyophilization) |
Cryoprotectants prevent vesicular fusion; dry powder rehydrates into intact vesicles in GI tract. |
Extends shelf-life; enables solid capsule formulation[15]. (Lacidipine) |
|
Limited cellular internalization |
Galactosylated Stearylamine |
Galactose actively binds ASGPRs on hepatocytes for receptor-mediated endocytosis. |
High liver specificity; avoids systemic distribution[33]. (Sofosbuvir) |
|
Food Matrix Interference |
PGA + Chitosan Coating |
PGA shields in stomach acid; shell expands in intestine to release cargo via mixed micelles. |
Delayed colonic delivery[31] (Trans-resveratrol) |
|
TRANSDERMAL & TOPICAL DELIVERY |
|||
|
Stratum corneum barrier |
PEGylated Edge Activators |
PEG hydrates & swells skin; creates ultra-deformable vesicles to squeeze through pores. |
Improves elasticity and permeation enhancement[27]. (Ketoconazole) |
|
Short dermal residence |
Bilosome-Loaded Hydrogels |
Polymeric gel network entraps vesicles, increasing skin hydration and providing a localized depot. |
Sustained transdermal delivery and prolonged flux[43]. (Dapagliflozin)
|
|
MUCOSAL DELIVERY |
|||
|
Nasolacrimal drainage |
Hyaluronic Acid (HA) Enrichment |
HA binds corneal mucin, forming a viscous, bio-adhesive interface. |
Sustained release; prolonged corneal retention[28]. (Felodipine) |
|
Rapid precorneal elimination |
Nano-Bilosomal Ocular Inserts |
Solid polymer insert slowly erodes in the conjunctival sac, steadily releasing intact vesicles. |
Sustained biphasic release kinetics; reduces dosing frequency[21]. (Voriconazole) |
|
Blood-brain barrier constraint |
Thermosensitive In Situ Gels |
Liquid formulation undergoes sol-gel transition at nasal temperature, preventing nasal drip. |
Bypasses the BBB via direct olfactory nerve transport[42]. (Glibenclamide) |
|
SYSTEMIC & TUMOR TARGETING |
|||
|
Pulmonary Clearance: Macrophage and mucociliary clearance |
Chitosan Nanoparticle Coating |
Charge-masking evades macrophages; chitosan opens tight junctions in alveolar epithelium. |
Enables deep alveolar deposition and protects payload[16]. (Daclatasvir) |
|
Tumour Targeting: Poor cellular uptake |
Folic Acid (FA)-PEG-Cholesterol |
Folic acid targets overexpressed cancer receptors; PEG extends blood circulation. |
Triggers active endocytosis and improves cytotoxicity[30]. (Vorinostat) |
|
Lipophilic instability in the tumour microenvironment. |
Zein Protein Stabilization |
Zein protein matrix coats the lipidic structure, preventing premature rupture in vivo. |
Stabilizes vesicle integrity and actively enhances cellular apoptosis[35]]. (Piceatannol) |
|
Inadequate tumour targeting |
Radio-iodination (131I) |
Radioactive tagging allows non-invasive tracking while vesicles exploit the leaky tumor vasculature (EPR effect). |
Enables real-time biodistribution tracking and targeted theranostics[40]. (Resveratrol) |
|
Poor accumulation in atherosclerotic lesions. |
Dextran Sulfate (DS) Coating |
DS binds SR-A scavenger receptors expressed on plaque-associated macrophages. |
Targeted stealth; lowers atherogenic index[13]. (Naringenin) |
CONCLUSION
Engineered bilosomes overcome the limitations of conventional vesicular carriers by using bile salts as membrane fluidizers. This makes the carriers both stable and flexible, allowing them to penetrate biological barriers. Bilosomes demonstrate multi-route versatility. They resist gastrointestinal degradation and enable oral lymphatic uptake, deform to travel the rigid transdermal stratum corneum, and reversibly modify tight junctions for efficient mucosal, ocular, and nose-to-brain delivery. Also, active surface modification allows systemic formulations to specifically target tumors and deep cellular compartments. Finally, the conversion of liquid dispersions into solid-state probilosomes addresses the challenge of long-term storage stability. By bridging the gap between supramolecular engineering and industrial scalability, modified bilosomes offer a highly adaptable platform for targeted, route-specific drug delivery.
REFERENCES







Nebin Koshy, Sudhir Mr, Arun Raj R, Akhila P, Anandhu Dineshan, Sreejith V, Sarath S, Engineered Bilosomes: Formulation Strategies, Surface Modifications, and Delivery Across Biological Barriers, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 2614-2629. https://doi.org/10.5281/zenodo.21340107
 10.5281/zenodo.21340107
10.5281/zenodo.21340107