
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Usha Dwarkadas Pathrikar Institute of Pharmacy, Dongargaon (Kawad), Phulambri, Chatrapati Sambhajinagar – 431111.
Paracetamol is widely used antipyretic and analgesic in pediatrics. Conventional liquid formulations suffer from limited stability, hydrolysis, and microbial contamination. This study developed paracetamol dry powder sachets for reconstitution into an oral suspension to improve stability, palatability, and dosing accuracy. A powder blend was prepared using suitable diluents, sweeteners, suspending agents, flavors, and preservatives. Pre-formulation studies included organoleptic evaluation, solubility, and drug–excipient compatibility. Flow properties were assessed via angle of repose, bulk density, tapped density, Carr’s index, and Hausner’s ratio. After reconstitution, the suspension was evaluated for pH, viscosity, sedimentation volume, redispersibility, drug content uniformity, and in vitro dissolution. The optimized formulation showed good flow, acceptable pH and viscosity, uniform drug content, and rapid drug release. Accelerated stability studies showed no significant changes in appearance or potency. These results indicate dry powder sachets are a stable, accurate, and patient-friendly alternative to conventional pediatric liquid paracetamol formulations
Paracetamol, also known as acetaminophen, is one of the most extensively used analgesic and antipyretic agents worldwide. It is widely recommended for the management of mild to moderate pain and fever, particularly in pediatric patients due to its favorable safety profile when administered at therapeutic doses[1,2] The World Health Organization includes paracetamol in its Model List of Essential Medicines for children, emphasizing its clinical importance in primary healthcare settings.[3]
Pediatric drug delivery presents unique challenges compared to adult formulations. Children, especially infants and young patients, often experience difficulty swallowing solid dosage forms such as tablets or capsules[4].As a result, liquid dosage forms including syrups and suspensions are generally preferred in pediatric therapy. These formulations allow flexible dosing based on body weight and provide ease of administration [5]. However, conventional liquid preparations are associated with several limitations, including reduced chemical stability, risk of microbial contamination, need for higher concentrations of preservatives, bulky packaging, and shorter shelf life [6].Paracetamol, being slightly soluble in water, and is commonly formulated as an oral suspension for pediatric use [7]. In ready-to-use suspensions, prolonged storage may lead to issues such as sedimentation, caking, degradation of the active pharmaceutical ingredient, and changes in viscosity [8] additionally, exposure to moisture and temperature variations can adversely affect the quality of the liquid formulation. These factors can compromise dose uniformity and therapeutic efficacy [9]
To overcome these limitations, dry powder formulations intended for reconstitution prior to administration have gained significant importance. Dry powder sachets for oral suspension offer improved chemical and physical stability by minimizing exposure to aqueous environments during storage[10].The product is reconstituted with a specified quantity of purified water immediately before use, thereby reducing degradation and enhancing shelf life[11] Furthermore, sachet packaging provides accurate unit dosing, ease of transportation, improved patient compliance, and better protection against environmental factors such as humidity and light[12]
The formulation of dry powder sachets requires careful selection of excipients to ensure proper flow characteristics, uniform drug distribution, and rapid reconstitution. Sweetening agents are incorporated to mask the bitter taste of paracetamol, while suspending agents help maintain uniform dispersion of drug particles after reconstitution [13] Preservatives may be included to ensure microbial safety during the in-use period. Optimization of these components is essential to achieve acceptable organoleptic properties, viscosity, and sedimentation behavior [14]
Evaluation of reconstitutable dry powder formulations involves comprehensive pre-formulation and post-reconstitution studies. Pre-formulation parameters such as solubility, compatibility, and flow properties play a crucial role in ensuring content uniformity and manufacturability[15] After reconstitution, the suspension must meet quality parameters including appropriate pH, viscosity, sedimentation volume, drug content, and dissolution profile[16] Stability studies conducted under recommended conditions provide assurance of product quality throughout its intended shelf life[17] In recent years, regulatory authorities such as the International Council for Harmonization of Technical Requirements for
Pharmaceuticals for Human Use have established guidelines to ensure the quality, safety, and efficacy of pharmaceutical formulations, including stability testing requirements. Compliance with these standards is essential for developing pediatric formulations suitable for clinical use [18]
Considering the widespread use of paracetamol in pediatric therapy and the limitations associated with conventional liquid preparations, the development of dry powder sachets for reconstitution represents a promising and practical approach. Therefore, the present study aims to formulate and evaluate paracetamol dry powder sachets intended for reconstitution into oral suspension, focusing on physicochemical properties, stability, and overall suitability for pediatric administration.
Fever and pain are among the most common clinical symptoms encountered in pediatric practice. Effective management of these conditions is essential to prevent complications such as dehydration, febrile seizures, discomfort, and poor feeding. Paracetamol remains the first-line drug of choice in children due to its established efficacy, minimal gastrointestinal irritation, and lower risk of adverse effects when compared with non-steroidal anti-inflammatory drugs. Its central inhibition of prostaglandin synthesis provides antipyretic and analgesic effects without significant anti-inflammatory action [21]
Despite its therapeutic advantages, the formulation of paracetamol for pediatric administration requires careful consideration of factors such as dose flexibility, palatability, safety, and stability. Children are particularly sensitive to taste, texture, and odor, which directly influence medication adherence. Bitter taste and unpleasant mouthfeel may result in refusal or incomplete dosing, ultimately affecting therapeutic outcomes. Therefore, pediatric formulations must incorporate suitable sweetening and flavoring agents to enhance acceptability without compromising stability
Another critical factor in pediatric dosage form design is dose accuracy. Since dosing in children is typically calculated based on body weight (mg/kg), formulations must allow precise measurement and uniform drug distribution. In conventional liquid suspensions, sedimentation of drug particles during storage may lead to inconsistent dosing if not adequately shaken before use. Improper redispersion can result in underdosing or overdosing, both of which may pose safety concerns [22]
Dry powder sachets designed for reconstitution provide a strategic solution to these challenges. By storing the active pharmaceutical ingredient in a dry state, the risk of hydrolytic degradation is significantly minimized. The absence of water during storage enhances chemical stability and reduces the likelihood of microbial growth. Upon addition of a measured quantity of water, the powder rapidly forms a homogeneous suspension suitable for immediate administration. This approach ensures both stability during shelf life and uniform dispersion at the time of use.[21] From a pharmaceutical technology perspective, the development of reconstitutable powder systems demands optimization of particle size distribution, flowability, and blending uniformity. Good flow properties are essential for accurate filling of sachets and maintaining content uniformity. The choice and concentration of suspending agents influence sedimentation rate, viscosity, and redispersibility of the final suspension. Excessive viscosity may hinder pourability, whereas insufficient viscosity may cause rapid sedimentation. Hence, a balance must be achieved to produce a stable and easily administrable formulation [22]
In addition to formulation considerations, packaging plays a vital role in product performance. Sachets made from moisture-resistant laminated materials provide protection against environmental humidity and oxygen exposure. Unit-dose packaging also improves portability, reduces contamination risk during repeated use, and simplifies administration in community or rural healthcare settings
Where refrigeration facilities may be limited. Regulatory expectations for pediatric formulations emphasize safety, quality, and performance consistency. Stability studies conducted under controlled temperature and humidity conditions help predict product shelf life and ensure compliance with established pharmaceutical standards. Evaluation of parameters such as drug content, pH, viscosity, sedimentation behavior, and dissolution characteristics confirms the suitability of the formulation for therapeutic use.[23]
In view of the increasing demand for stable, patient-friendly, and accurate pediatric dosage forms, the formulation of paracetamol dry powder sachets for reconstitution into oral suspension represents a rational and innovative approach. The present research is therefore undertaken to design, develop, and systematically evaluate such a formulation with the objective of improving stability, ensuring dose precision, and enhancing pediatric patient compliance [24]
OBJECTIVE
1. To formulate paracetamol dry powder sachets intended for reconstitution into oral suspension for pediatric use.
2. To enhance the stability of paracetamol by developing a dry dosage form with improved shelf life.
3. To select and optimize suitable excipients for taste masking and improved palatability.
4. To evaluate drug–excipient compatibility using appropriate analytical techniques[14]
5. To assess pre-compression parameters such as angle of repose, bulk
Density, tapped density, Carr’s index, and Hausner’s ratio [22]
6. To ensure uniform blending and content uniformity of the dry powder formulation.
7. To evaluate post-reconstitution parameters including
pH, viscosity, sedimentation volume, and redispersibility [13]
8. To determine drug content and ensure compliance with pharmacopeial limits.
9. To perform in vitro dissolution studies to evaluate the drug release profile.
10. To conduct stability studies under accelerated conditions to assess physical stability, chemical stability, and shelf life of the formulation.
DRUG PROFILE (Paracetamol) [1, 7]
1) General Information
• Other Name: Acetaminophen
• Chemical Name: N-(4-hydroxyphenyl) acetamide
• Category: Analgesic and Antipyretic
• Official in: IP, BP, US
2) Chemical information [1, 3]
• Molecular Formula: C₈H₉NO₂
• Molecular Weight: 151.16 g/mol
• Structural Formula:
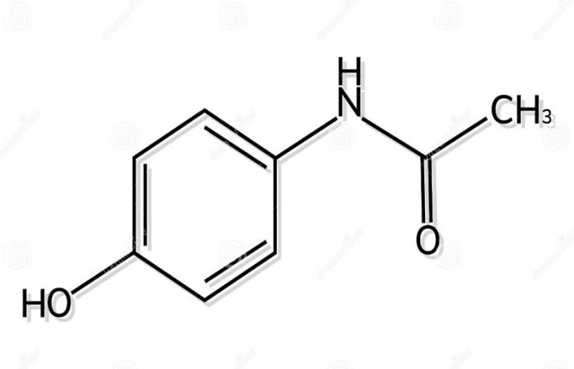
Figure 1: Structure of Paracetamol(26)
• Functional Groups: Phenolic hydroxyl group and amide group
• Melting Point: 168–172°C
• pKa: ~9.5
3) Physical properties
• Appearance: White or almost white crystalline powder
• Odor: Odorless
• Taste: Slightly bitter
• Solubility:
Slightly soluble in cold water
Freely soluble in alcohol
Soluble in acetone
• Stability: Stable under normal conditions but may degrade in the presence of
moisture and heat
4) Pharmacological Classification:
• Paracetamol belongs to the class of centrally acting analgesic and antipyretic
drugs. It has minimal anti-inflammatory activity compared to NSAIDs.
• According to the World Health Organization, paracetamol is included in the
Model List of Essential Medicines for children.
6. Mechanism of Action : [2,19]
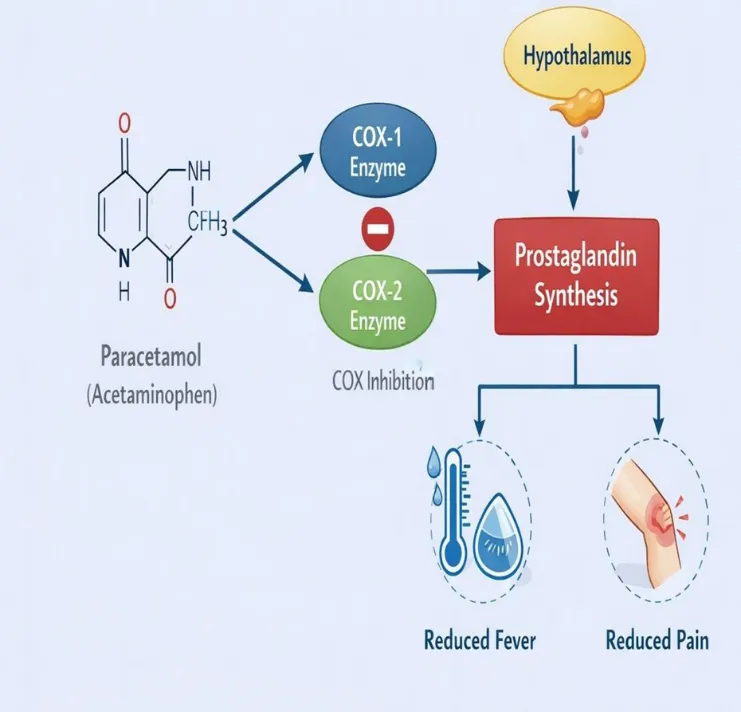
Figure 2: Mechanism of action
1. Paracetamol Administration
After oral administration, paracetamol is absorbed into the bloodstream and crosses the blood–brain barrier to act primarily in the central nervous system (CNS).
2. Inhibition of COX Enzymes
In the brain, paracetamol inhibits cyclooxygenase (COX) enzymes, mainly COX-2 and to a lesser extent COX-1. These enzymes are responsible for converting arachidonic acid into prostaglandins.
3. Reduced Prostaglandin Synthesis
By inhibiting COX enzymes, paracetamol decreases the production of prostaglandins in the CNS. Prostaglandins are chemical mediators involved in the transmission of pain signals and regulation of body temperature.
4. Action on Hypothalamus (Fever Reduction)
In cases of fever, pyrogens stimulate prostaglandin production in the hypothalamus, which raises the body’s temperature set point.
Reduced prostaglandin levels reset the hypothalamic thermostat to normal, leading to:
• Vasodilation
• Sweating
• Heat loss
→ Result: Reduction of fever
5. Pain Relief Mechanism
Prostaglandins sensitize pain receptors (nociceptors) in the CNS. Lower prostaglandin levels decrease pain signal transmission, resulting in: → Reduced perception of pain
6. Minimal Peripheral Anti-inflammatory Effect
Unlike NSAIDs, paracetamol has very weak inhibition of peripheral COX enzymes. Therefore, it does not significantly reduce inflammation or affect platelet aggregation.
7. Pharmacokinetics[1.2] Absorption
• Rapidly and almost completely absorbed from the gastrointestinal tract.
• Peak plasma concentration: 30–60 minutes after oral administration.
Distribution
• Widely distributed in body fluids.
• Plasma protein binding: 10–25%.
Metabolism
• Primarily metabolized in the liver by conjugation with glucuronic acid and sulfate.
• A small fraction is metabolized via cytochrome P450 to form a toxic metabolite (NAPQI), which is detoxified by glutathione.
Elimination
• Excreted mainly in urine as conjugated metabolites.
• Half-life: 2–3 hours (may vary in neonates and hepatic impairment).
8. Pediatric Dos
• 10–15 mg/kg every 4–6 hours
• Maximum daily dose: 60–75 mg/kg/day (depending on guidelines)
9. Adverse Effects
a) Rare and mil
b) Hepatotoxicity
c) Nausea and vomiting
d) Liver failure (in severe cases)
MATERIALS AND METHODS
Active Pharmaceutical Ingredient (API)
• Paracetamol
Excipients
• Sucrose (fine powder) – sweetening agent, diluent
• Sorbitol (fine powder) – sweetener, humectant, anti-caking agent
• Sucralose (fine powder) – high-intensity sweetener
• Lemon-lime flavour (spray-dried) – flavouring agent
• Purified water – vehicle for reconstitution
Method of Preparation:
The dry powder blend was prepared using the direct dry mixing method as described by Kafedjiiski et al and modified for paracetamol [22, 13]
1. Sieving: All ingredients were accurately weighed and passed through a #100 mesh sieve.
2. Pre-mix Preparation: A pre-mix was prepared by geometrically diluting the low-dose ingredients (Sucralose, lemon-lime flavour) with approximately 100 mg of Sorbitol.
3. Addition of Remaining Bulking Agents: The remaining Sorbitol and all Sucrose were added to the pre-mix and blended thoroughly in a polythene bag for 10 minutes.
4. Geometric Addition of Paracetamol: Paracetamol was added in three portions, blending thoroughly after each addition.
5. Final Blending: The final blend was mixed by mortal pestle for 15-20 minutes.
Sachet Filling: The final blend was filled into tri-laminate foil sachets to achieve the target weight of approximately 2.0 g per sachet. Sachets were heat-sealed
EVALUATION PARAMETER
Pre-Formulation Studies [14]
Pre-formulation studies are conducted to understand the physicochemical properties of the drug and to ensure compatibility with excipients before formulation development.
1. Organoleptic Properties
Objective: To evaluate the physical and sensory characteristics of the drug. Parameters Assessed:
• Color
• Odor
• Taste
• Physical appearance
Method: The drug sample was visually examined under normal lighting conditions. Taste and odor were evaluated carefully.
2. Solubility Study
Objective: To determine solubility behavior of the drug in different solvents.
Method:
• Excess drug was added to solvents (distilled water, ethanol, buffer pH 1.2 and 6.8).
• The mixture was shaken for 24 hours at room temperature.
• Filtered and analyzed spectrophotometrically.
3. Melting Point Determination
Objective: To confirm purity and identity of drug.
Method:
• Capillary method was used.
• Small amount of drug was filled in sealed capillary tube.
• Temperature at which drug melted was recorded
Pre-Compression Parameters [22]
Pre-compression studies evaluate powder blend properties before filling into sachets.
1. Angle of Repose
Objective: To determine the flowability of the powder blend. Method (Funnel Method):
• A funnel was fixed at a certain height.
• Powder blend was allowed to flow freely onto a flat surface forming a cone.
• Height (h) and radius (r) of the cone were measured. Formula:
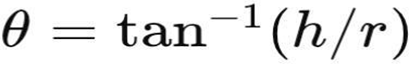
Interpretation:
Angle of Repose (°) Flow Property
< 25° Excellent
25–30° Good
30–40° Passable
> 40° Poor
2. Bulk Density
Objective: To determine the packing ability of powder under loose conditions.
Method:
• Accurately weighed powder was transferred into a graduated cylinder.
• Initial volume (V₀) was noted
• Formula:
3. Tapped Density
Objective: To determine the packing ability after mechanical tapping. Method:
• The cylinder containing powder was tapped mechanically (500– 1000 taps).
• Final tapped volume (Vt) was recorded. Formula:
4. Carr’s Compressibility Index
Objective: To evaluate compressibility and flow property of powder. Formula:
Interpretation:
Carr’s Index (%) Flow Property
5–15 Excellent
16–20 Good
21–25 Fair
>25 Poor
5. Hausner’s Ratio
Objective: To determine interparticle friction and flow behavior. Formula:
Interpretation:
Hausner’s Ratio Flow Property
1.00–1.11 Excellent
1.12–1.18 Good
Post-Reconstitution Evaluation [15,25]
1. Appearance
Objective: To check homogeneity and absence of lumps. Observation: Color, clarity, uniformity, absence of caking.
2. pH Measurement
Objective: To determine acidity/alkalinity of suspension. Method:
• Measured using calibrated digital pH meter.
3. Viscosity (Brookfield Viscometer) Objective: To measure thickness of suspension. Method:
• Measured using Brookfield viscometer at specific rpm.
• Appropriate spindle selected.
4. Sedimentation Volume
Objective: To evaluate physical stability.
Method:
• Suspension stored in graduated cylinder.
• Sediment volume recorded after specific time.
5. Formula:
Where:
Vu = Ultimate sediment volume
Vo = Original volume
6. Redispersibility :
Objective: To check ease of redistributing sediment.
Method: Number of manual inversions required to redisperse sediment
recorded.
7. Stability Studies[17,24]
Objective: To determine shelf life and stability. Conditions:
• 25°C ± 2°C / 60% RH ± 5% RH
• 40°C ± 2°C / 75% RH ± 5% RH
Parameters Evaluated:
• Physical appearance
• Drug content
• pH
• Dissolution
8. In-Vitro Dissolution Study
Objective: To determine drug release profile. Method:
a. USP Dissolution Apparatus II (Paddle method).
b. Temperature maintained at 37 ± 0.5°C.
c. Samples withdrawn at predetermined intervals.
d. Analyzed using UV spectrophotometer
RESULT AND DISCUSSION:
Pre-Formulation Studies
1. Organoleptic Properties
• Color: White crystalline powder
• Odor: Odorless
• Taste: Slightly bitter
• Appearance: Fine, free-flowing powder
2. Solubility Study
• Distilled Water - Slightly soluble
• Ethanol - Freely soluble
• pH 1.2 buffer – Soluble
• pH 6.8 - buffer
• Moderately soluble
3. Melting Point
Observed melting point: 168–170°C
Pre-Compression Parameters
|
Sr.no |
Parameter |
Result |
Interpretation |
|
1 |
Angle of Repose |
27.5° |
Good flow property |
|
2 |
Bulk Density |
0.52 g/cm³ |
Acceptable packing ability |
|
3 |
Tapped Density |
0.61 g/cm³ |
Suitable compressibility |
|
4 |
Carr’s Index |
14.75% |
Good flowability (11–15%) |
|
5 |
Hausner’s Ratio |
1.17 |
Good flow (<1.25) |
Post-Reconstitution Evaluation
|
Sr.no |
Parameter |
Result |
Interpretation |
|
1 |
ppearance |
Smooth, uniform suspension, no lumps |
Physically stable |
|
2 |
pH |
5.8 |
Within acceptable range (4.5–6.5) |
|
3 |
Viscosity |
10 cps (Spindle 3, 60 rpm) |
Adequate pourability & stability |
|
4 |
Sedimentation Volume (F) |
0.92 |
High stability |
|
5 |
Redispersibility |
1–3 inversions |
Easily redispersible |
|
6 |
Drug Content (243 nm) |
87% |
Within limit (95– 105%) |
CONCLUSION
Paracetamol dry powder sachets were successfully formulated and evaluated. The optimized formulation showed good flow properties, acceptable reconstitution characteristics, satisfactory drug release, and stability. This dosage form is suitable for pediatric administration due to improved stability, accurate dosing, and better patient compliance.
REFERENCES

R. Daud, Shreyas Gore, Krushna Tarte, Datta Wahatule, Priti Jadhav, Formulation And Evaluation of Paracetamol Dry Powder Sachets for Reconstitution for Pediatric Use, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 2454-2463, https://doi.org/10.5281/zenodo.20613441
 10.5281/zenodo.20613441
10.5281/zenodo.20613441