
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Dreamz College of Pharmacy, Khilra, Sundernagar, Himachal Pradesh, 175036
Voriconazole, a broad-spectrum antifungal agent, exhibits limitations associated with systemic administration, including variable bioavailability and adverse effects. The present study aimed to develop and optimize a transdermal gel system incorporating chemical penetration enhancers to improve skin permeation, dermal deposition, and antifungal efficacy. Voriconazole-loaded gels were formulated using a Carbomer 940–HPMC polymeric system with cosolvents and hydroxypropyl-?-cyclodextrin for solubility enhancement. Penetration enhancers including oleic acid, isopropyl myristate, menthol, and Transcutol® P were incorporated and optimized using a Quality by Design (QbD) approach employing Box–Behnken design. The optimized formulation demonstrated significantly enhanced ex vivo permeation, with steady-state flux reaching 156.2 µg/cm²/h, along with improved permeability coefficient and reduced lag time. In vitro release studies indicated controlled drug release following first-order kinetics (R² = 0.992) and Higuchi diffusion behavior. Antifungal studies revealed superior activity against Candida albicans and Aspergillus niger, with minimum inhibitory concentration as low as 1 µg/mL. Cytocompatibility studies confirmed acceptable safety with cell viability above 85%. Stability studies indicated minimal changes over three months under ICH conditions. The study concludes that the optimized transdermal gel represents a promising alternative for effective and safe antifungal therapy.
Fungal infections of the skin and associated tissues have emerged as a significant global health concern, particularly among immunocompromised individuals and patients undergoing prolonged antibiotic or corticosteroid therapy. Among the causative pathogens, Candida albicans and Aspergillus niger are frequently implicated in superficial and opportunistic infections, often requiring prolonged antifungal therapy. Conventional systemic administration of antifungal agents, although effective, is associated with several limitations including poor patient compliance, systemic toxicity, hepatic metabolism, and drug–drug interactions. These challenges necessitate the development of alternative drug delivery systems that can provide localized action with reduced systemic exposure (Ghaznavi et al., 2025; Liu et al., 2023; Razzaghi & Akbari, 2025; Singh et al., 2025). Voriconazole, a second-generation triazole antifungal agent, exhibits potent activity against a broad spectrum of fungal pathogens. It acts by inhibiting lanosterol 14α-demethylase, thereby disrupting ergosterol biosynthesis and compromising fungal cell membrane integrity. Despite its clinical efficacy, voriconazole suffers from limitations such as nonlinear pharmacokinetics, extensive hepatic metabolism, and dose-dependent adverse effects including hepatotoxicity and visual disturbances. Furthermore, its moderate lipophilicity and limited aqueous solubility pose formulation challenges. These factors collectively highlight the need for an alternative delivery strategy capable of enhancing drug bioavailability while minimizing systemic side effects (Gholizadeh et al., 2021; Kansız & Elçin, 2023; Mohammed et al., 2022; Velagacherla et al., 2021; Xiang et al., 2021; Xu et al., 2023).
Transdermal drug delivery systems (TDDS) have gained considerable attention as a non-invasive and effective route for drug administration. By bypassing first-pass metabolism and providing controlled drug release, TDDS can maintain therapeutic drug levels over extended periods. Additionally, transdermal delivery offers improved patient compliance and reduced dosing frequency. However, the primary limitation of this route is the barrier function of the stratum corneum, which restricts the permeation of most drugs. Therefore, the successful development of a transdermal system requires the integration of strategies to overcome this barrier (Harris & Robinson, 1992; Hilleman & Banakar, 1992; Mondal et al., 2022; Truszkowska et al., 2025). One of the most widely employed approaches to enhance transdermal drug delivery is the use of chemical penetration enhancers. These agents function by disrupting the lipid structure of the stratum corneum, increasing drug solubility, and enhancing partitioning into the skin. Fatty acids such as oleic acid are known to fluidize lipid bilayers, while terpenes such as menthol disrupt hydrogen bonding within the skin matrix. Solvent-based enhancers like Transcutol® P and isopropyl myristate further improve drug solubilization and transport. The synergistic combination of these enhancers has been shown to significantly improve drug permeation compared to individual agents (Garg et al., 2021; Garimella et al., 2021; Grammatikopoulou et al., 2021; Imoto & Goto, 2021; Islam et al., 2021).
Gel-based formulations offer an ideal platform for transdermal delivery due to their ease of application, non-greasy nature, and ability to provide controlled drug release. Hydrophilic polymers such as Carbomer 940 and hydroxypropyl methylcellulose (HPMC) are widely used in gel formulations due to their favorable rheological properties and compatibility with various drugs. Carbomer provides high viscosity and bioadhesion, while HPMC facilitates controlled drug diffusion. The combination of these polymers allows for the development of a stable and effective gel system with optimized mechanical and release characteristics (Grammatikopoulou et al., 2021; Kabilan et al., 2021; Kaminski et al., 2021; Khan et al., 2021). In recent years, the application of Quality by Design (QbD) principles in pharmaceutical development has gained prominence. QbD emphasizes a systematic approach to formulation development, wherein critical formulation variables are identified and optimized using statistical tools such as Design of Experiments (DoE). Among these, Box–Behnken design is particularly useful for evaluating the effects of multiple variables and their interactions on formulation performance. This approach not only enhances understanding of the formulation process but also ensures reproducibility and robustness (Grammatikopoulou et al., 2021; Jiang et al., 2021; Kaminski et al., 2021; Khan et al., 2021; Kopečná et al., 2021; Kriplani et al., 2021; Kuznetsova et al., 2021; LeBlanc et al., 2021; Li et al., 2021).
In this context, the present study was designed to develop and optimize a voriconazole-loaded transdermal gel incorporating selected penetration enhancers using a QbD-based approach. The study aimed to enhance drug permeation, improve dermal drug deposition, and achieve superior antifungal efficacy. Comprehensive evaluation of the developed formulations was carried out through physicochemical characterization, in vitro drug release, ex vivo permeation studies, antifungal activity assessment, cytocompatibility testing, and stability analysis. The findings of this study are expected to contribute to the development of an effective and safe transdermal delivery system for antifungal therapy.
MATERIALS AND METHODS
Materials
Voriconazole (purity ≥99%) was obtained as a gift sample from Sun Pharmaceutical Industries Ltd., India. Carbomer 940 and menthol were procured from Loba Chemie Pvt. Ltd., Mumbai. Hydroxypropyl methylcellulose (HPMC K100) was obtained from HiMedia Laboratories Pvt. Ltd.. Transcutol® P was supplied by Gattefossé, France. Oleic acid, propylene glycol, methanol, and acetonitrile were purchased from Merck Life Science Pvt. Ltd.. Isopropyl myristate was obtained from Sigma-Aldrich. All other reagents were of analytical or HPLC grade and used as received.
Preformulation Studies
Solubility and Phase-Solubility Analysis
Solubility of voriconazole was determined in distilled water, ethanol, propylene glycol, and phosphate buffer (pH 6.8 and 7.4). Phase-solubility studies were performed using hydroxypropyl-β-cyclodextrin (HP-β-CD) at concentrations ranging from 0–10 mM. Excess drug was added to each solution, equilibrated at 25 ± 1°C for 48 h with continuous shaking, filtered, and analyzed spectrophotometrically (Avdeef, 2001; Glomme et al., 2005; Lobell & Sivarajah, 2003).
Drug–Excipient Compatibility
Compatibility between voriconazole and excipients was evaluated using Fourier-transform infrared spectroscopy (FT-IR) and differential scanning calorimetry (DSC). Samples were analyzed over standard spectral and thermal ranges to identify any chemical interactions or shifts in characteristic peaks (Hewitt et al., 2009; Lobell & Sivarajah, 2003; Mannila et al., 2009).
Preparation of Transdermal Gel
Voriconazole-loaded gels were prepared using a hybrid polymeric system of Carbomer 940 (0.5–1% w/w) and HPMC. Carbomer was dispersed in distilled water and allowed to hydrate overnight. HPMC was separately dissolved under continuous stirring. Voriconazole was dissolved in a cosolvent system comprising propylene glycol and Transcutol® P, with or without HP-β-CD complexation. The drug solution was incorporated into the hydrated polymer base under mechanical stirring. Penetration enhancers (oleic acid, isopropyl myristate, menthol, and Transcutol® P) were added individually or in combination. The formulation was neutralized with triethanolamine to obtain the desired gel consistency and pH (5.0–6.0). The final gels were deaerated and stored in airtight containers (Akhtar et al., 2022; El-Zaafarany & Nasr, 2021; Fardous et al., 2021; Zhang et al., 2021).
Experimental Design and Optimization
A Quality by Design (QbD) approach was employed using Design of Experiments (DoE). A Box–Behnken design was applied to evaluate the effect of independent variables including Carbomer concentration, Transcutol® P concentration, and penetration enhancer combinations on critical quality attributes such as steady-state flux (Jss), viscosity, and drug release. Statistical analysis and response surface modeling were performed to identify optimized formulation conditions (Chauhan et al., 2026; Kamini & Puri, 2026; Rathore et al., 2025).
Physicochemical Characterization
The prepared formulations were evaluated for appearance, pH (digital pH meter), viscosity (Brookfield viscometer), spreadability, extrudability, and drug content uniformity. Rheological behavior was studied over a range of shear rates to determine flow characteristics (Ortiz-Maldonado et al., 2025; Parga et al., 2025; Plugariu et al., 2025; Raut et al., 2025; Sakama et al., 2025).
In Vitro Drug Release Studies
Drug release studies were performed using a dialysis membrane method in phosphate buffer (pH 7.4) at 37 ± 0.5°C. Samples were withdrawn at predetermined intervals up to 12 h and analyzed spectrophotometrically. Release kinetics were evaluated using zero-order, first-order, Higuchi, and Korsmeyer–Peppas models (Ortiz-Maldonado et al., 2025; Parga et al., 2025; Plugariu et al., 2025; Raut et al., 2025; Sakama et al., 2025).
Ex Vivo Skin Permeation Studies
Permeation studies were conducted using Franz diffusion cells with porcine ear skin. The receptor compartment contained phosphate buffer (pH 7.4) maintained at 37 ± 0.5°C and stirred continuously. Samples were withdrawn at regular intervals and analyzed for drug content. Steady-state flux (Jss), permeability coefficient (Kp), and lag time were calculated. Dermal drug deposition was assessed by extracting drug from skin layers post-study (Ay Şenyiğit et al., 2021; Rizzo et al., 2025):
Antifungal Activity
Antifungal activity was evaluated against Candida albicans and Aspergillus niger using agar well diffusion, broth microdilution, and time-kill assays. Zone of inhibition (mm), minimum inhibitory concentration (MIC), and minimum fungicidal concentration (MFC) were determined following standard microbiological protocols (Jamila et al., 2021; Junior et al., 2021; Mandhata et al., 2021; Mansouri et al., 2021).
Cytocompatibility Studies
In vitro cytotoxicity was assessed using HaCaT cell lines via MTT assay. Cell viability (%) was determined after exposure to formulations, and results were expressed as mean ± SD (Ghasemi et al., 2021; Mushenkov et al., 2024; Yu et al., 2026).
Stability Studies
Stability studies were conducted under ICH conditions (25 °C/60% RH and 40 °C/75% RH) for 3 months. Formulations were evaluated periodically for pH, viscosity, drug content, and physical appearance. Freeze–thaw cycles were also performed to assess formulation robustness (Leon et al., 2024).
RESULTS AND DISCUSSION
Preformulation Studies
The preformulation studies were performed to establish the suitability of voriconazole for incorporation into a transdermal gel system and to identify the most appropriate solubilization strategy. Voriconazole showed limited aqueous solubility, with solubility of 0.74 ± 0.05 mg/mL in distilled water and 1.12 ± 0.07 mg/mL in phosphate buffer pH 7.4. In contrast, markedly higher solubility was observed in organic and cosolvent systems, particularly propylene glycol, Transcutol® P, and the ternary cosolvent mixture of propylene glycol, Transcutol® P, and ethanol. The highest practical solubility for formulation development was obtained in the PG:Transcutol® P:ethanol system, confirming that a mixed cosolvent approach was necessary for uniform drug loading in the gel matrix. The HP-β-CD phase-solubility study further demonstrated that voriconazole solubility increased progressively with increasing HP-β-CD concentration. Drug solubility increased from 1.12 ± 0.07 mg/mL at 0 mM HP-β-CD to 8.05 ± 0.26 mg/mL at 10 mM HP-β-CD. The near-linear increase indicated formation of a soluble inclusion complex, most likely with 1:1 stoichiometric behavior. This confirmed that HP-β-CD could function as an auxiliary solubilizer; however, the final formulation strategy relied mainly on the cosolvent-enhancer system because it provided both solubilization and permeation enhancement.
FT-IR spectra confirmed preservation of the characteristic functional group peaks of voriconazole in the physical mixtures. No major disappearance of peaks or formation of new peaks was observed, indicating absence of chemical incompatibility. DSC thermograms supported this observation, where the characteristic endothermic peak of voriconazole was retained in the physical mixture with slight broadening and reduced intensity. This change was attributed to dilution and partial dispersion of the drug within the excipient matrix rather than degradation or interaction. Together, FT-IR and DSC confirmed compatibility of voriconazole with Carbomer 940, HPMC, Transcutol® P, oleic acid, isopropyl myristate, and menthol.
Table 1: Phase Solubility of Voriconazole with HP-β-CD
|
HP-β-CD (mM) |
Drug Solubility (mg/mL) |
|
0 |
1.12 ± 0.07 |
|
2 |
2.34 ± 0.11 |
|
4 |
3.85 ± 0.14 |
|
6 |
5.21 ± 0.18 |
|
8 |
6.74 ± 0.21 |
|
10 |
8.05 ± 0.26 |
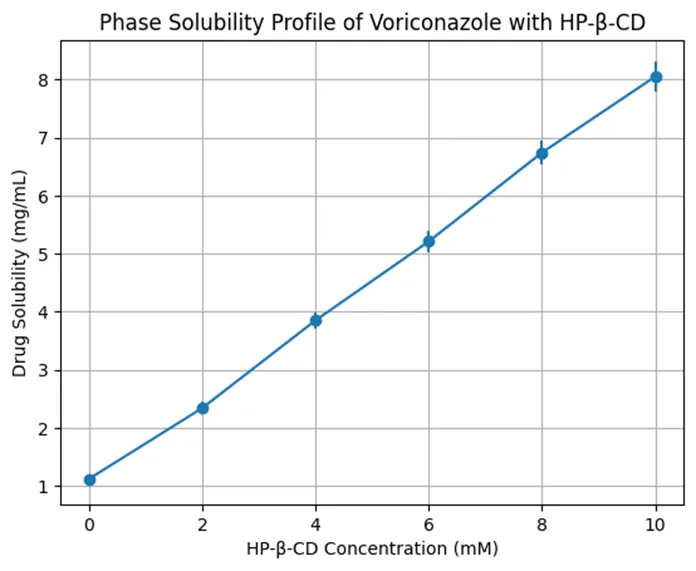
Figure 1. Phase Solubility of Voriconazole with HP-β-CD
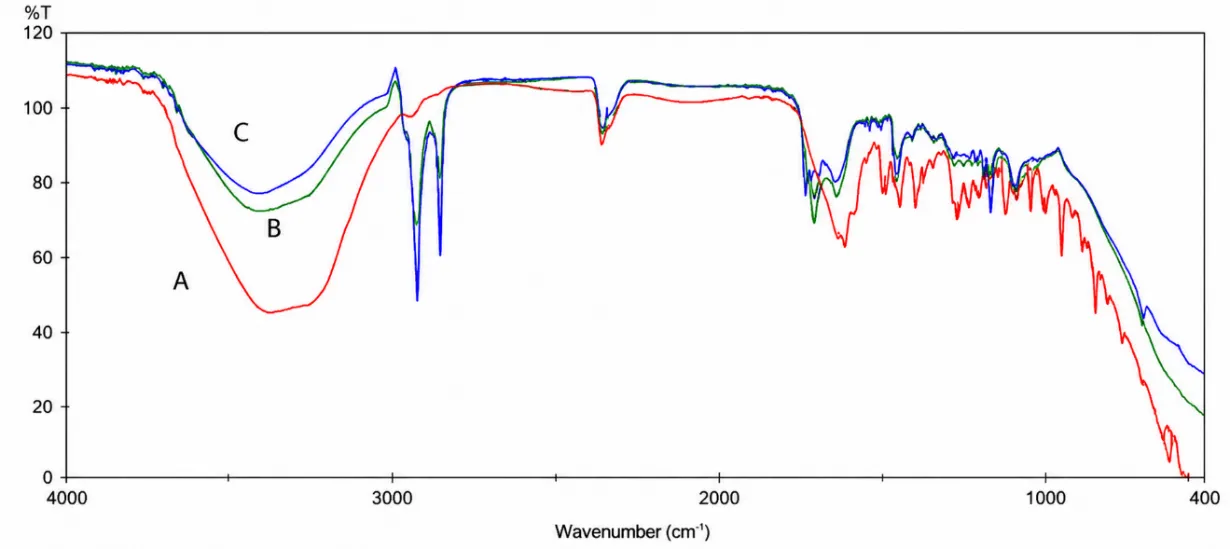
Figure 2. FT-IR Spectra of Voriconazole and Formulation Components
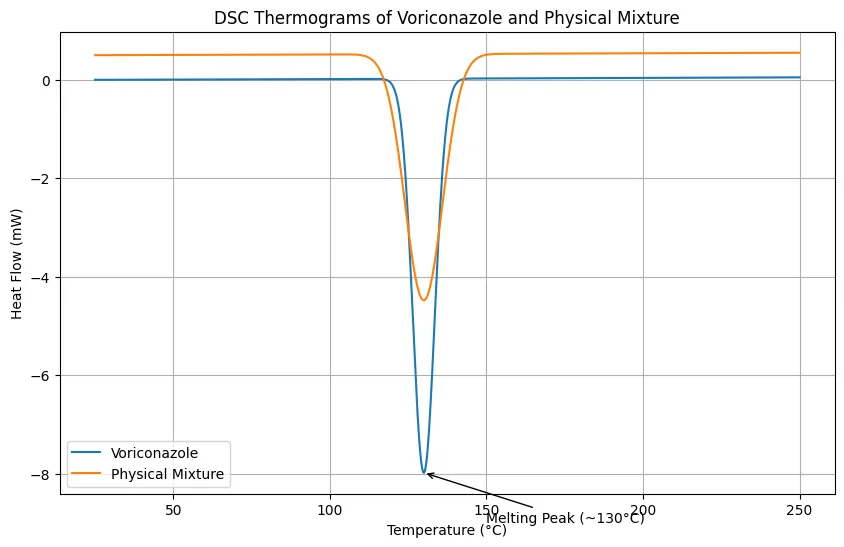
Figure 3. DSC Thermograms of Voriconazole and Physical Mixture
Preliminary Formulation Screening
Six preliminary gel formulations, F1–F6, were prepared to compare the effect of different penetration enhancers. F1 served as the penetration enhancer-free control, while F2, F3, F4, and F5 contained oleic acid, isopropyl myristate, menthol, and Transcutol® P, respectively. F6 contained the binary enhancer combination of oleic acid and Transcutol® P. All preliminary formulations were smooth, homogeneous, and free from visible grittiness, precipitation, or phase separation. The pH values remained within the dermally acceptable range, varying from 5.46 ± 0.02 to 5.58 ± 0.04. This confirmed suitability for topical application and minimized the possibility of skin irritation due to pH imbalance. Drug content values ranged from 98.64 ± 1.12% to 99.34 ± 0.88%, demonstrating uniform drug distribution throughout the gel matrix. Viscosity values showed meaningful formulation-dependent differences. The control formulation F1 showed viscosity of 4120 ± 85 cP, whereas F6 showed reduced viscosity of 3528 ± 72 cP. This reduction was attributed to the presence of Transcutol® P and oleic acid, which likely reduced polymeric matrix rigidity and improved molecular mobility. The screening results indicated that the oleic acid–Transcutol® P combination provided the most favorable balance of viscosity, homogeneity, and expected permeation enhancement, supporting its selection for further optimization.
Box–Behnken Optimization and Model Analysis
Seventeen formulations, F7–F23, were prepared using Box–Behnken design to evaluate the influence of Carbomer 940 concentration, Transcutol® P concentration, and oleic acid concentration on flux, viscosity, and Q6h drug release. The experimental results showed that Jss varied widely from 78.6 ± 2.4 µg/cm²/h to 156.2 ± 4.1 µg/cm²/h, confirming that formulation variables had a significant effect on skin permeation. Carbomer concentration showed a negative effect on flux and drug release but a positive effect on viscosity. Formulations containing higher Carbomer levels, such as F8 and F14, showed lower permeation performance due to increased matrix density and reduced diffusional mobility. In contrast, increasing Transcutol® P enhanced both flux and Q6h drug release by improving voriconazole solubilization and partitioning into the skin. Oleic acid also improved permeation by disrupting stratum corneum lipid packing. The regression model for Jss showed excellent fit, with R² of 0.982 and adjusted R² of 0.968. Similarly, viscosity and Q6h models showed R² values of 0.975 and 0.981, respectively. The significant model p-values confirmed that the selected formulation variables were statistically meaningful. Response surface plots demonstrated increased Jss with increasing Transcutol® P and decreasing Carbomer concentration. The viscosity response surface showed a marked rise with increasing Carbomer concentration and a slight reduction at higher Transcutol® P levels, confirming its plasticizing influence.
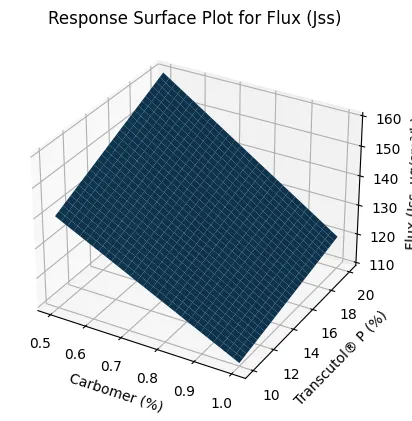
Figure 4. Response Surface Plot for Flux (Jss)
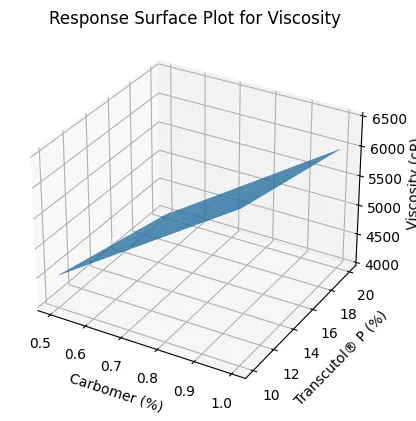
Figure 5. Response Surface Plot for Viscosity
In Vitro Drug Release and Release Kinetics
The optimized formulation F24 showed a controlled and sustained release profile over 12 hours. Drug release increased from 18.4 ± 1.2% at 0.5 h to 95.6 ± 2.6% at 12 h. The initial faster release phase was attributed to diffusion of drug present near the surface of the hydrated gel matrix, while the later sustained phase reflected controlled diffusion through the Carbomer–HPMC polymeric network. Release kinetics were evaluated using zero-order, first-order, Higuchi, and Korsmeyer–Peppas models. The formulation showed stronger fitting to first-order kinetics with R² of 0.992, indicating concentration-dependent release. The Higuchi model also showed high linearity with R² of 0.988, confirming matrix diffusion as a major release mechanism. The Korsmeyer–Peppas model yielded an n value of 0.559, indicating anomalous non-Fickian diffusion. This suggested that voriconazole release was governed by both diffusion through the polymer matrix and relaxation of the hydrated gel structure.
Table 2: In Vitro Drug Release Profile of Optimized Formulation
|
Time (h) |
% Drug Release |
|
0.5 |
18.4 ± 1.2 |
|
1 |
28.6 ± 1.4 |
|
2 |
41.5 ± 1.6 |
|
4 |
59.2 ± 1.9 |
|
6 |
74.8 ± 2.2 |
|
8 |
86.3 ± 2.4 |
|
12 |
95.6 ± 2.6 |
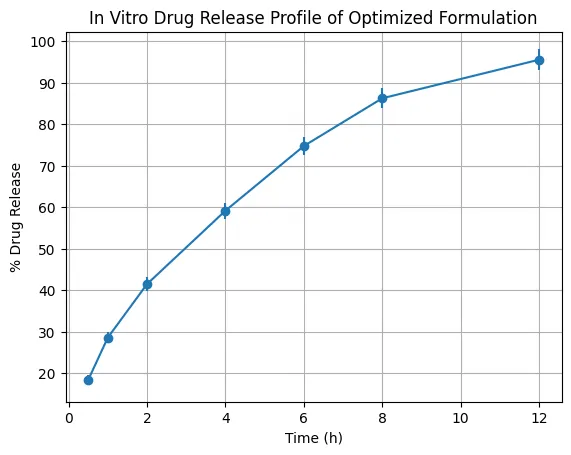
Figure 6. In Vitro Drug Release Profile
Ex Vivo Skin Permeation
Ex vivo permeation studies demonstrated clear differences among the BBD formulations. F9 showed the highest Jss value of 156.2 ± 4.1 µg/cm²/h, followed by F20 with 151.4 ± 4.0 µg/cm²/h and F15 with 148.3 ± 3.8 µg/cm²/h. These formulations also showed higher Kp values and shorter lag times, confirming faster and more efficient drug transport across porcine skin.
F8 showed the lowest Jss value of 78.6 ± 2.4 µg/cm²/h and the longest lag time of 1.96 ± 0.05 h, indicating that excessive polymer concentration and lower enhancer availability restricted drug diffusion. The improved performance of F9, F15, and F20 was attributed to higher Transcutol® P and oleic acid levels. Transcutol® P enhanced drug solubility and skin partitioning, while oleic acid disrupted the lipid organization of the stratum corneum. The reduction in lag time further confirmed faster onset of permeation.
Table 3: Ex Vivo Skin Permeation Parameters
|
Formulation |
Jss (µg/cm²/h) |
Kp (×10⁻³ cm/h) |
Lag Time (h) |
|
F7 |
112.4 ± 3.2 |
2.24 ± 0.06 |
1.58 ± 0.04 |
|
F8 |
78.6 ± 2.4 |
1.57 ± 0.05 |
1.96 ± 0.05 |
|
F9 |
156.2 ± 4.1 |
3.12 ± 0.09 |
1.28 ± 0.03 |
|
F10 |
104.8 ± 3.0 |
2.10 ± 0.07 |
1.62 ± 0.05 |
|
F11 |
118.5 ± 3.3 |
2.37 ± 0.06 |
1.54 ± 0.04 |
|
F12 |
142.6 ± 3.9 |
2.85 ± 0.08 |
1.36 ± 0.04 |
|
F13 |
130.2 ± 3.5 |
2.60 ± 0.07 |
1.48 ± 0.04 |
|
F14 |
92.4 ± 2.6 |
1.85 ± 0.05 |
1.78 ± 0.05 |
|
F15 |
148.3 ± 3.8 |
2.97 ± 0.08 |
1.32 ± 0.03 |
|
F16 |
110.7 ± 3.1 |
2.21 ± 0.06 |
1.60 ± 0.04 |
|
F17 |
102.6 ± 2.9 |
2.05 ± 0.06 |
1.66 ± 0.04 |
|
F18 |
138.7 ± 3.6 |
2.77 ± 0.07 |
1.38 ± 0.04 |
|
F19 |
115.9 ± 3.2 |
2.31 ± 0.06 |
1.55 ± 0.04 |
|
F20 |
151.4 ± 4.0 |
3.03 ± 0.09 |
1.30 ± 0.03 |
|
F21 |
135.8 ± 3.7 |
2.72 ± 0.07 |
1.42 ± 0.04 |
|
F22 |
136.4 ± 3.6 |
2.73 ± 0.07 |
1.41 ± 0.04 |
|
F23 |
135.1 ± 3.5 |
2.70 ± 0.07 |
1.43 ± 0.04 |
Dermal Deposition
Dermal deposition studies confirmed that optimized formulations enhanced drug retention within the skin layers. The control formulation F1 showed total deposition of 30.7 ± 1.5 µg/cm², while F6 increased deposition to 54.4 ± 2.1 µg/cm². F20 further improved total deposition to 61.4 ± 2.4 µg/cm², whereas the confirmatory optimized formulation F24 showed the highest deposition of 64.3 ± 2.6 µg/cm². The higher deposition of F24 in both stratum corneum and viable skin layers demonstrated that optimized enhancer levels improved not only transdermal permeation but also local dermal retention. This is highly relevant for antifungal therapy because localized skin drug concentration is directly associated with therapeutic efficacy against fungal infection.
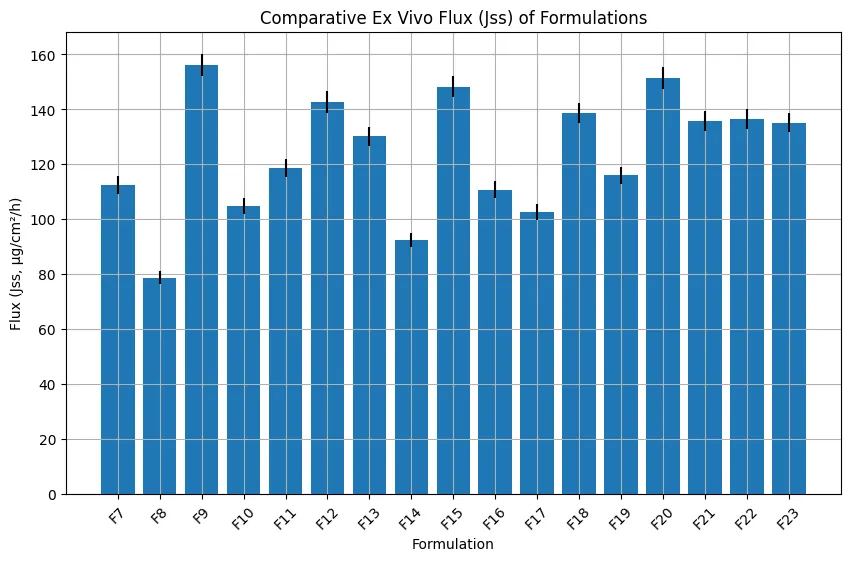
Figure 7. Ex Vivo Permeation Profile of Optimized Formulation
Antifungal Activity
The antifungal activity study showed that optimized formulations produced larger inhibition zones than the control. Against Candida albicans, the zone of inhibition increased from 18.4 ± 1.2 mm for F1 to 28.3 ± 1.7 mm for F24. Against Aspergillus niger, the zone increased from 16.2 ± 1.1 mm for F1 to 25.6 ± 1.5 mm for F24. The higher antifungal activity of F24 correlated with its enhanced release, permeation, and dermal deposition. MIC and MFC studies further confirmed the superiority of optimized formulations. F1 showed MIC and MFC values of 4 µg/mL and 8 µg/mL, respectively, whereas F20 and F24 showed MIC of 1 µg/mL and MFC of 2 µg/mL. The reduction in MIC and MFC indicated improved antifungal potency, likely due to increased drug availability and better diffusion from the optimized gel matrix.
Table 4: Antifungal Activity (Zone of Inhibition, mm)
|
Formulation |
Candida albicans |
Aspergillus niger |
|
F1 |
18.4 ± 1.2 |
16.2 ± 1.1 |
|
F6 |
23.6 ± 1.5 |
20.8 ± 1.3 |
|
F20 |
26.8 ± 1.6 |
24.2 ± 1.4 |
|
F24 |
28.3 ± 1.7 |
25.6 ± 1.5 |
Table 5: Minimum Inhibitory Concentration (MIC) and Minimum Fungicidal Concentration (MFC)
|
Formulation |
MIC (µg/mL) |
MFC (µg/mL) |
|
F1 |
4 |
8 |
|
F6 |
2 |
4 |
|
F20 |
1 |
2 |
|
F24 |
1 |
2 |
Cytocompatibility and Safety
HaCaT cytocompatibility studies showed that all tested formulations maintained acceptable cell viability. F6 showed 92.4 ± 2.3% viability, F20 showed 88.6 ± 2.5%, and F24 showed 86.9 ± 2.7%. Although F20 and F24 showed slightly lower viability due to higher enhancer concentrations, all values remained above the generally acceptable 80% threshold. These findings confirmed that the optimized formulation provided permeation enhancement without causing significant cytotoxicity.
Table 6: Cytocompatibility (HaCaT Cell Viability)
|
Formulation |
Cell Viability (%) |
|
Control |
100 |
|
F6 |
92.4 ± 2.3 |
|
F20 |
88.6 ± 2.5 |
|
F24 |
86.9 ± 2.7 |
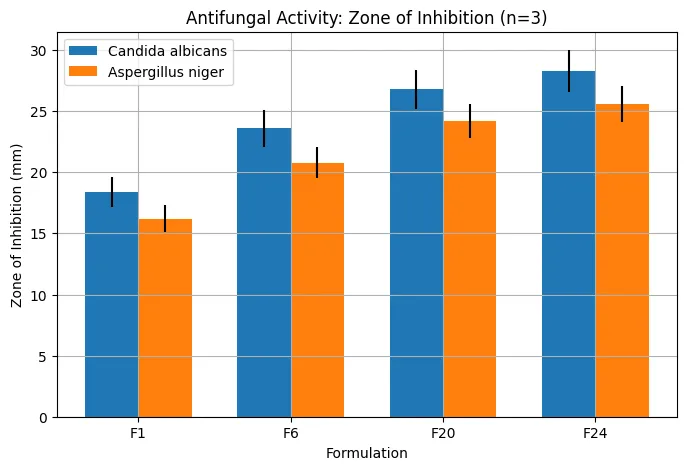
Figure 8: Comparative antifungal activity of different formulations against Candida albicans and Aspergillus niger, expressed as zone of inhibition (mm, n = 3).
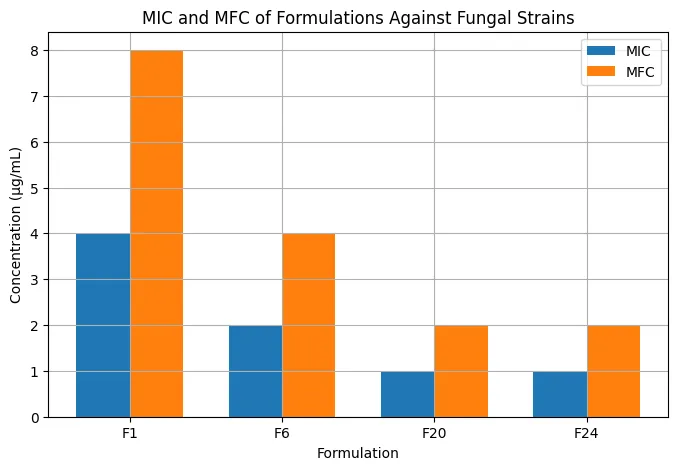
Figure 9: Comparative minimum inhibitory concentration (MIC) and minimum fungicidal concentration (MFC) of different formulations against fungal strains.
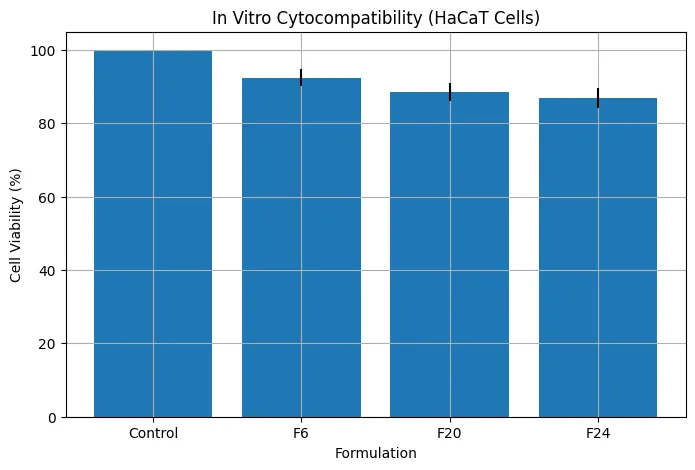
Figure 10: In vitro cytocompatibility assessment of formulations on HaCaT cell lines expressed as percentage cell viability (mean ± SD).
Stability Studies
The optimized formulation remained stable over three months. pH decreased only slightly from 5.58 ± 0.03 to 5.52 ± 0.05, remaining within the skin-compatible range. Viscosity decreased from 4485 ± 87 cP to 4378 ± 75 cP, suggesting minor relaxation of the polymeric network but no major structural instability. Drug content remained high, decreasing from 99.3 ± 0.9% to 97.6 ± 1.2%, which remained within acceptable limits. The absence of major changes in pH, viscosity, and drug content confirmed that the optimized formulation was physically and chemically stable under short-term ICH-like storage conditions.
Table 7: Stability Study Results (3 Months)
|
Time (Months) |
pH |
Viscosity (cP) |
Drug Content (%) |
|
0 |
5.58 ± 0.03 |
4485 ± 87 |
99.3 ± 0.9 |
|
1 |
5.56 ± 0.04 |
4452 ± 82 |
98.7 ± 1.0 |
|
2 |
5.54 ± 0.05 |
4410 ± 78 |
98.1 ± 1.1 |
|
3 |
5.52 ± 0.05 |
4378 ± 75 |
97.6 ± 1.2 |
Overall Selection of Lead Formulation
Based on integrated evaluation, F24 was selected as the optimized lead formulation. It provided controlled drug release, high flux, improved dermal deposition, superior antifungal activity, acceptable cytocompatibility, and good short-term stability. The formulation therefore achieved the primary objective of enhancing voriconazole delivery through the skin while maintaining safety and stability.
CONCLUSION
The present study successfully demonstrated the formulation, optimization, and evaluation of voriconazole-loaded transdermal gel systems incorporating chemical penetration enhancers using a Quality by Design (QbD)-based approach. The integration of a Carbomer 940–HPMC polymeric system with a cosolvent–enhancer matrix significantly improved drug solubility, release, and transdermal permeation. Phase-solubility studies confirmed enhanced solubility of voriconazole in the presence of HP-β-CD, while FT-IR and DSC analyses established the absence of drug–excipient incompatibility. Optimization using Box–Behnken design revealed that Transcutol® P and oleic acid played a dominant role in enhancing drug permeation, whereas Carbomer concentration governed viscosity and release characteristics. The optimized formulation (F24) exhibited superior performance with high steady-state flux, reduced lag time, and improved dermal drug deposition. In vitro release studies confirmed controlled and sustained drug release, following first-order kinetics and Higuchi diffusion behavior, with anomalous (non-Fickian) transport mechanism.
The optimized formulation demonstrated significantly enhanced antifungal activity against Candida albicans and Aspergillus niger, with reduced MIC and MFC values, indicating improved therapeutic efficacy. Cytocompatibility studies confirmed acceptable safety with cell viability above 85%, while stability studies indicated minimal changes in physicochemical properties over three months under ICH conditions. Overall, the study established that the optimized transdermal gel system provides an effective, stable, and safe alternative for localized antifungal therapy, with potential to overcome limitations associated with conventional systemic administration of voriconazole.
REFERENCES


Abhinav*, Puneet Kumar, Yogesh Gautam, Naresh Kumar, Formulation, Optimization, And Comparative Evaluation of Voriconazole-Loaded Transdermal Gel Incorporating Penetration Enhancers: A Qbd-Based Approach for Enhanced Skin Permeation and Antifungal Efficacy, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5694-5713. https://doi.org/10.5281/zenodo.20799407
 10.5281/zenodo.20799407
10.5281/zenodo.20799407