
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Guru Nanak College of Pharmaceutical Sciences, Dehradun
Generic drugs play a vital role in modern healthcare systems by providing cost-effective alternatives to innovator or branded medicines while maintaining comparable quality, safety, and efficacy. The development of generic pharmaceutical products has expanded significantly due to increasing healthcare costs, patent expirations of blockbuster drugs, and the growing demand for affordable treatment options worldwide. Generic drug development primarily focuses on demonstrating pharmaceutical equivalence and bioequivalence with the reference listed drug (RLD), rather than repeating extensive preclinical and clinical studies conducted for the innovator product. The process of generic drug development involves several critical stages, including selection of the reference product, preformulation studies, formulation optimization, analytical method development, stability studies, scale-up, and regulatory submission. Among these, bioequivalence studies are considered the cornerstone of generic drug approval because they establish that the generic product exhibits similar rate and extent of drug absorption as the branded counterpart. Pharmacokinetic parameters such as maximum plasma concentration (Cmax), time to reach maximum concentration (Tmax), and area under the plasma concentration-time curve (AUC) are commonly evaluated to assess bioequivalence. Regulatory agencies such as the USFDA, EMA, CDSCO, and WHO have established stringent guidelines and acceptance criteria to ensure therapeutic equivalence and patient safety. In recent years, advancements such as Quality by Design (QbD), Biopharmaceutics Classification System (BCS)-based biowaivers, in-vitro dissolution modeling, and artificial intelligence-driven pharmaceutical development have significantly improved the efficiency and reliability of generic drug development. Despite these advances, several challenges remain, including development of complex generics, modified-release formulations, and compliance with evolving global regulatory requirements. This review highlights the principles, regulatory requirements, formulation strategies, and bioequivalence assessment methods involved in generic drug development. It also discusses current challenges, recent technological advancements, and future perspectives in the field. The article emphasizes the importance of robust bioequivalence studies in ensuring therapeutic interchangeability and promoting global access to affordable medicines.
Generic drugs have become one of the most significant components of the modern pharmaceutical industry due to their ability to provide safe, effective, and affordable treatment options to patients worldwide. A generic drug is a pharmaceutical product that contains the same active pharmaceutical ingredient (API), dosage form, strength, route of administration, and therapeutic effect as an already approved innovator or branded drug. These medicines are introduced into the market after the expiration of patent protection and exclusivity rights of the original product. Unlike innovator drugs, generic medicines do not require extensive preclinical and clinical trials for approval because their safety and efficacy have already been established by the reference listed drug (RLD). Instead, manufacturers are primarily required to demonstrate pharmaceutical equivalence and bioequivalence with the branded product. Bioequivalence studies ensure that the generic formulation delivers the drug into systemic circulation at a similar rate and extent as the reference product, thereby confirming therapeutic equivalence. Due to stringent regulatory requirements imposed by authorities such as the USFDA, EMA, WHO, and CDSCO, generic medicines are considered interchangeable with branded products in clinical practice. The growing acceptance of generic drugs among healthcare professionals and patients has contributed substantially to reducing healthcare expenditure while maintaining treatment quality and accessibility [1].
The importance of generic medicines in healthcare systems has increased dramatically over the past few decades, especially in developing and low-income countries where access to expensive branded medicines is limited. Rising healthcare costs, increasing prevalence of chronic diseases, and expanding global populations have placed enormous pressure on healthcare infrastructures, making affordable medicines a necessity rather than an option. Generic drugs play a crucial role in improving patient access to essential medicines for conditions such as hypertension, diabetes, cardiovascular disorders, infectious diseases, and cancer [2]. Since generic products are generally marketed at significantly lower prices than innovator drugs, they reduce the financial burden on both patients and healthcare systems. In many countries, generic substitution policies and insurance reimbursement programs actively encourage the use of generics to control pharmaceutical expenditures. Furthermore, the widespread availability of generic medicines promotes medication adherence because patients are more likely to continue therapy when treatment costs are affordable. This ultimately contributes to better disease management, reduced hospitalization rates, and improved public health outcomes [3].
In addition to economic benefits, generic medicines also stimulate competition within the pharmaceutical market. The entry of multiple generic manufacturers following patent expiry leads to price reductions and encourages innovation among pharmaceutical companies. Innovator firms are often motivated to develop new drug delivery systems, improved formulations, and novel therapeutic agents to maintain market competitiveness. Consequently, the generic pharmaceutical sector indirectly drives scientific advancement and continuous pharmaceutical innovation. Moreover, the generic drug industry contributes significantly to the global economy by generating employment opportunities in manufacturing, research and development, regulatory affairs, quality assurance, clinical research, and supply chain management. Countries such as India have emerged as major global suppliers of generic medicines due to their strong manufacturing infrastructure, skilled workforce, and cost-effective production capabilities. India is often referred to as the “pharmacy of the world” because of its extensive contribution to affordable medicines across numerous countries, particularly in the supply of antiretroviral drugs, antibiotics, and essential medications during public health emergencies [4-10].
The evolution of the generic pharmaceutical industry has been shaped by several regulatory, economic, and technological developments over the years. Initially, the pharmaceutical market was dominated by innovator companies that held exclusive rights to manufacture and market patented drugs. Before the establishment of modern regulatory frameworks, generic medicines often faced skepticism regarding their quality, safety, and therapeutic performance. However, the introduction of stringent regulatory guidelines significantly improved confidence in generic products. One of the most influential milestones in the development of the generic industry was the enactment of the Hatch–Waxman Act in the United States in 1984. This legislation established the Abbreviated New Drug Application (ANDA) pathway, allowing generic manufacturers to seek approval by demonstrating bioequivalence rather than repeating costly clinical trials. The Hatch–Waxman Act created a balance between encouraging pharmaceutical innovation and promoting the availability of affordable generic medicines. Following this regulatory advancement, the global generic pharmaceutical market expanded rapidly, leading to increased competition and wider patient access to medicines [11].
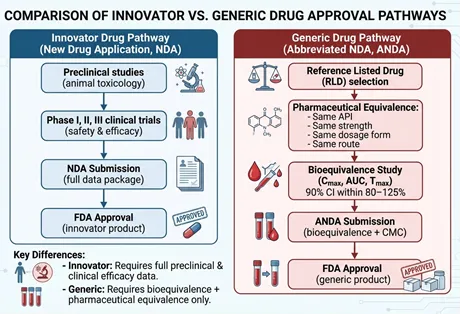
Fig: 1 Regulatory Pathway for Generic Drug Approval – ANDA System
Technological advancements have further accelerated the growth and sophistication of generic drug development. Modern pharmaceutical technologies such as Quality by Design (QbD), process analytical technology (PAT), advanced analytical instruments, and computer-assisted formulation development have improved the efficiency, quality, and reproducibility of generic products. In addition, the implementation of Good Manufacturing Practices (GMP), Good Laboratory Practices (GLP), and Good Clinical Practices (GCP) has strengthened quality assurance throughout the product lifecycle. The introduction of Biopharmaceutics Classification System (BCS)-based biowaivers has simplified approval procedures for certain drug categories by reducing the need for in-vivo bioequivalence studies under specific conditions. Furthermore, advances in bioanalytical methods such as high-performance liquid chromatography (HPLC) and liquid chromatography–mass spectrometry (LC-MS/MS) have enhanced the accuracy and reliability of pharmacokinetic analysis in bioequivalence studies [12].
Despite remarkable progress, the generic pharmaceutical industry continues to face several challenges, including patent litigation, development of complex generics, regulatory harmonization issues, and increasing demands for quality compliance. Complex dosage forms such as liposomal formulations, inhalation products, transdermal systems, and biologics require sophisticated manufacturing technologies and extensive characterization studies. Additionally, global regulatory agencies are continuously updating guidelines to ensure patient safety and product quality, requiring manufacturers to adapt to evolving standards. Nevertheless, the future of the generic pharmaceutical industry remains highly promising due to increasing patent expirations, rising demand for affordable healthcare, and ongoing advancements in pharmaceutical sciences. The continued expansion of generic medicines is expected to play a pivotal role in achieving equitable healthcare access and improving therapeutic outcomes worldwide [13].
Regulatory Framework for Generic Drug Approval
The regulatory framework for generic drug approval plays a critical role in ensuring that generic medicines are safe, effective, high in quality, and therapeutically equivalent to their innovator counterparts. Generic drug approval systems have been developed by regulatory authorities worldwide to facilitate the availability of affordable medicines while maintaining strict standards for pharmaceutical quality and patient safety. Unlike innovator drugs, generic medicines are not required to undergo extensive preclinical and clinical efficacy studies because the safety and effectiveness of the active pharmaceutical ingredient (API) have already been established through the approval of the reference listed drug (RLD). Instead, generic manufacturers must demonstrate pharmaceutical equivalence and bioequivalence with the innovator product. Regulatory agencies establish comprehensive guidelines regarding formulation development, manufacturing practices, analytical validation, stability testing, labeling, and bioequivalence assessment to ensure therapeutic interchangeability between generic and branded medicines. These frameworks also help maintain public confidence in generic products and support global healthcare systems by reducing medication costs and increasing accessibility [14].
Several national and international regulatory authorities oversee the approval and monitoring of generic medicines. Among the most influential agencies are the United States Food and Drug Administration (USFDA), European Medicines Agency (EMA), Central Drugs Standard Control Organization (CDSCO), and World Health Organization (WHO). These organizations establish scientific and regulatory standards for drug approval, manufacturing quality, pharmacovigilance, and post-marketing surveillance. The USFDA regulates generic drugs in the United States through stringent requirements under the Federal Food, Drug, and Cosmetic Act. Similarly, the EMA coordinates the scientific evaluation and approval of medicines within the European Union, while CDSCO governs pharmaceutical approvals and quality standards in India. The WHO plays a major role in promoting harmonized international standards, particularly for developing countries, through technical guidelines and prequalification programs. In addition to these organizations, other regulatory bodies such as the Medicines and Healthcare products Regulatory Agency (MHRA) in the United Kingdom and the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan contribute significantly to global pharmaceutical regulation. Despite variations in regional requirements, most regulatory frameworks share common objectives related to safety, efficacy, quality assurance, and bioequivalence demonstration [15].
One of the most important regulatory pathways for generic drug approval is the Abbreviated New Drug Application (ANDA) system established in the United States. The ANDA pathway allows generic manufacturers to obtain approval without repeating extensive animal and human clinical trials required for innovator drugs. Instead, the applicant must demonstrate that the generic product is pharmaceutically equivalent and bioequivalent to the reference listed drug. Pharmaceutical equivalence requires the generic product to contain the same active ingredient, dosage form, route of administration, strength, and labeling characteristics as the branded drug. Bioequivalence studies compare pharmacokinetic parameters such as maximum plasma concentration (Cmax), time to reach maximum concentration (Tmax), and area under the plasma concentration-time curve (AUC) between the test and reference formulations. Acceptance criteria generally require the 90% confidence interval for the ratio of these parameters to fall within the range of 80–125% [16-20].
The ANDA process significantly reduces the time and cost associated with drug approval, thereby encouraging market competition and increasing patient access to affordable medicines. In addition to bioequivalence data, applicants must submit detailed information regarding manufacturing processes, analytical validation, stability studies, packaging, labeling, and compliance with Good Manufacturing Practices (GMP).
The development of the modern generic pharmaceutical industry was greatly influenced by the Hatch–Waxman Act, officially known as the Drug Price Competition and Patent Term Restoration Act of 1984. This landmark legislation transformed the pharmaceutical market in the United States by balancing the interests of innovator companies and generic manufacturers. Before the enactment of this law, generic drug approval was often complicated, time-consuming, and financially burdensome because manufacturers were required to repeat extensive clinical studies. The Hatch–Waxman Act introduced the ANDA pathway, which allowed generic manufacturers to rely on the safety and efficacy data of the innovator drug while demonstrating bioequivalence. At the same time, the Act provided patent term restoration and market exclusivity benefits to innovator companies to compensate for the time lost during the regulatory approval process. The legislation also established the Paragraph I, II, III, and IV certification system related to patent status. Among these, Paragraph IV certification became highly significant because it allowed generic companies to challenge patents they considered invalid or non-infringed. The first generic applicant filing a successful Paragraph IV certification may receive 180 days of marketing exclusivity, which serves as a major economic incentive for early generic competition. The Hatch–Waxman Act is considered one of the most important regulatory reforms in pharmaceutical history because it accelerated generic drug availability while continuing to encourage pharmaceutical innovation [21].
Different regulatory agencies have established specific requirements for generic drug approval based on regional healthcare policies and scientific standards. The USFDA requires submission of ANDA applications supported by bioequivalence studies, chemistry-manufacturing-control (CMC) documentation, stability data, and GMP compliance. The EMA emphasizes comparative quality studies, bioequivalence evaluation, and risk management plans in accordance with European guidelines. In India, CDSCO regulates generic medicines under the Drugs and Cosmetics Act and mandates bioequivalence studies for many categories of drugs, especially for export-oriented and critical formulations. The WHO supports global harmonization by issuing technical guidelines related to generic product development, bioequivalence testing, stability studies, and pharmaceutical quality systems. WHO prequalification programs are especially important for medicines supplied to international health programs and low-income countries. Regulatory agencies also emphasize post-marketing surveillance and pharmacovigilance activities to monitor adverse drug reactions and ensure continued product safety after approval [22].
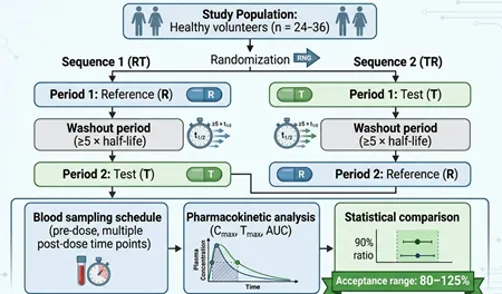
Fig: 2 Design of a Two‑Period, Two‑Sequence Crossover Bioequivalence Study
Patent expiry and market exclusivity are fundamental concepts governing the introduction of generic medicines into the pharmaceutical market. Pharmaceutical patents provide innovator companies with exclusive marketing rights for a specific period, usually 20 years from the date of patent filing. These patents protect intellectual property and allow innovator companies to recover the substantial investments associated with drug discovery, research, development, and clinical trials. During the exclusivity period, generic manufacturers are prohibited from marketing equivalent products. However, once the patent and regulatory exclusivity periods expire, generic companies can apply for approval and enter the market with therapeutically equivalent products. In some cases, generic manufacturers may challenge existing patents through Paragraph IV certification if they believe the patent is invalid, unenforceable, or not infringed by the generic formulation. Patent expiration often leads to intense market competition and substantial reductions in drug prices, benefiting healthcare systems and patients. Consequently, the balance between patent protection and generic competition remains a crucial aspect of pharmaceutical regulation and public health policy worldwide [23].
Drug Development Process for Generic Medicines
The development process for generic medicines is a highly systematic and scientifically driven procedure aimed at producing pharmaceutical products that are therapeutically equivalent to innovator or branded drugs. Generic drug development focuses on ensuring that the final product demonstrates comparable quality, safety, efficacy, and performance to the reference listed drug (RLD) while maintaining cost-effectiveness and regulatory compliance. Unlike innovator drug development, which involves extensive drug discovery and clinical research, generic drug development primarily emphasizes formulation optimization, pharmaceutical equivalence, and bioequivalence studies. The process includes several interconnected stages such as selection of the reference listed drug, preformulation studies, active pharmaceutical ingredient (API) characterization, compatibility testing, formulation development, excipient selection, manufacturing process optimization, scale-up, and technology transfer. Each stage is essential for achieving consistent product quality, regulatory approval, and successful commercialization [24].
The first and one of the most important steps in generic drug development is the selection of the Reference Listed Drug (RLD). The RLD is the innovator or branded product approved by regulatory authorities against which the generic product is compared. Regulatory agencies such as the United States Food and Drug Administration publish lists of approved reference products that can be used for generic drug development. Selection of the appropriate RLD is critical because the generic product must match the reference product in dosage form, strength, route of administration, labeling, and intended use. Developers carefully study the physicochemical properties, dissolution profile, packaging characteristics, and pharmacokinetic behavior of the RLD to design an equivalent generic formulation. Reverse engineering techniques are often used to understand the composition and manufacturing characteristics of the innovator product. Factors such as market demand, patent status, regulatory pathways, therapeutic significance, and commercial potential are also considered during RLD selection. A detailed understanding of the reference product provides the foundation for successful formulation development and bioequivalence achievement [25].
Following RLD selection, preformulation studies are conducted to evaluate the physicochemical and biopharmaceutical properties of the active pharmaceutical ingredient. Preformulation studies are essential because they provide scientific information required for selecting suitable formulation strategies and manufacturing processes. Important parameters evaluated during preformulation include solubility, dissolution rate, particle size distribution, polymorphism, pKa, partition coefficient, hygroscopicity, melting point, stability, and flow properties. These studies help determine how the API behaves under different environmental and processing conditions. For example, poorly water-soluble drugs may require particle size reduction, solid dispersion techniques, or use of solubilizing agents to improve dissolution and bioavailability. Similarly, APIs sensitive to moisture, heat, or light may require specialized manufacturing and packaging conditions to maintain stability. Preformulation studies also help identify potential challenges related to compressibility, flowability, and compatibility with excipients. Comprehensive preformulation analysis enables scientists to develop robust formulations capable of delivering consistent therapeutic performance [26].
API characterization and compatibility studies represent another crucial phase of generic drug development. Characterization of the active pharmaceutical ingredient involves detailed analysis of its chemical structure, purity, crystalline form, residual solvents, and impurity profile using advanced analytical techniques such as Fourier-transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), X-ray diffraction (XRD), high-performance liquid chromatography (HPLC), and mass spectrometry. These studies ensure that the API meets pharmacopeial and regulatory quality standards. Compatibility studies are conducted to evaluate possible interactions between the API and excipients used in the formulation. Drug–excipient interactions can significantly affect product stability, dissolution, efficacy, and shelf life. Compatibility testing is typically performed under accelerated storage conditions using analytical methods to detect physical or chemical changes in the formulation components. Incompatibilities such as discoloration, degradation, moisture absorption, or reduced potency may indicate the need for alternative excipients or formulation approaches. Compatibility studies are therefore essential for ensuring formulation stability and long-term product performance [27].
Formulation development is the core stage of generic drug development and involves designing a pharmaceutical product that closely mimics the innovator drug in terms of performance and quality attributes. Scientists aim to develop formulations with similar dissolution profiles, release characteristics, appearance, and stability as the reference product. Depending on the dosage form, formulation development may involve optimization of granulation techniques, compression parameters, coating processes, or modified-release mechanisms. For oral solid dosage forms such as tablets and capsules, factors including tablet hardness, friability, disintegration time, and dissolution behavior are carefully optimized. Formulation scientists conduct multiple experimental trials using different combinations of excipients and processing conditions to achieve the desired product characteristics. Comparative dissolution studies between the test and reference products are performed throughout development to evaluate similarity in drug release patterns. Advanced approaches such as Quality by Design (QbD) and Design of Experiments (DoE) are increasingly used to identify critical formulation variables and establish robust manufacturing processes. These approaches improve product consistency, reduce variability, and facilitate regulatory compliance [27-30].
Selection of excipients is another vital component of generic formulation development because excipients significantly influence product stability, manufacturability, and bioavailability. Excipients are inactive substances used to support drug formulation and may include diluents, binders, disintegrants, lubricants, glidants, coatings, preservatives, and coloring agents. Although excipients are pharmacologically inactive, they can affect dissolution, absorption, and overall therapeutic performance of the drug product. Therefore, careful selection of excipients is necessary to ensure compatibility with the API and achieve desired formulation characteristics. Generic manufacturers often attempt to use excipients similar to those present in the reference product to minimize formulation differences. Regulatory agencies also evaluate excipient safety and functionality during product approval. Factors such as regulatory acceptance, cost, availability, functionality, and manufacturing suitability are considered during excipient selection. For modified-release formulations, specialized polymers and release-controlling agents are selected to replicate the release behavior of the innovator product accurately [31].
Manufacturing process optimization is an essential stage aimed at developing a reproducible and efficient production process capable of consistently producing high-quality products. During this phase, process parameters such as blending time, granulation conditions, drying temperature, compression force, coating variables, and mixing speed are optimized to ensure uniformity and product performance. Process optimization minimizes variability and helps identify critical process parameters that influence product quality. Statistical and scientific tools such as Design of Experiments (DoE), process analytical technology (PAT), and risk assessment methods are widely used to establish process control strategies. Manufacturing optimization also includes evaluation of in-process controls, equipment performance, and environmental conditions to ensure compliance with Good Manufacturing Practices (GMP). Robust process development reduces batch failures, improves production efficiency, and enhances product reproducibility during large-scale manufacturing [32].
After successful laboratory-scale development and optimization, the formulation undergoes scale-up and technology transfer. Scale-up involves increasing production from pilot-scale batches to commercial manufacturing levels while maintaining product quality and consistency. This stage is challenging because changes in equipment size, mixing efficiency, heat transfer, and processing conditions can affect product characteristics. Scientists carefully evaluate process reproducibility and make necessary adjustments to maintain equivalence between pilot and commercial batches. Process validation studies are conducted to demonstrate that the manufacturing process consistently produces products meeting predetermined quality specifications. Technology transfer refers to the systematic transfer of product knowledge, manufacturing procedures, analytical methods, and quality requirements from the research and development unit to the commercial manufacturing site. Effective technology transfer requires detailed documentation, standard operating procedures, training programs, and coordination between development and production teams. Regulatory agencies require comprehensive evidence demonstrating that the scaled-up manufacturing process maintains the same quality, stability, and bioequivalence characteristics established during development. [33]
Overall, the drug development process for generic medicines is a multidisciplinary and highly regulated procedure involving pharmaceutical sciences, analytical chemistry, manufacturing technology, quality assurance, and regulatory affairs. Each stage, from RLD selection to commercial-scale production, contributes to the development of safe, effective, and affordable generic medicines that meet global regulatory standards. Continuous advancements in formulation science, analytical technologies, and process optimization strategies are further improving the efficiency and reliability of generic drug development, ultimately supporting broader patient access to high-quality medicines worldwide [34].
Quality by Design (QbD) in Generic Drug Development
Quality by Design (QbD) has emerged as one of the most important scientific and regulatory approaches in modern pharmaceutical development, particularly in the field of generic drug formulation and manufacturing. QbD is a systematic, science-based, and risk-oriented approach that emphasizes designing quality into pharmaceutical products rather than relying solely on end-product testing. The concept was introduced and strongly encouraged by the United States Food and Drug Administration and further supported through guidelines developed by the International Council for Harmonisation (ICH), especially ICH Q8, Q9, and Q10. In generic drug development, QbD helps manufacturers achieve consistent product quality, improved manufacturing efficiency, reduced variability, and enhanced regulatory flexibility. Unlike conventional pharmaceutical development, which primarily focuses on empirical testing and fixed manufacturing conditions, QbD aims to build a thorough understanding of the relationship between formulation components, manufacturing processes, and final product performance. This approach enables manufacturers to identify critical variables affecting product quality and establish scientifically justified control strategies throughout the product lifecycle [35].
The fundamental principle of QbD is that quality should be intentionally designed into the product from the initial stages of development. The QbD framework begins with defining the Quality Target Product Profile (QTPP), which outlines the desired characteristics of the final pharmaceutical product, including dosage form, strength, route of administration, dissolution behavior, pharmacokinetic properties, stability, and therapeutic performance. The QTPP serves as the foundation for formulation and process development. Once the QTPP is established, scientists identify Critical Quality Attributes (CQAs), which are physical, chemical, biological, or microbiological properties that must remain within predefined limits to ensure product safety, efficacy, and quality. Examples of CQAs for oral solid dosage forms include assay, content uniformity, dissolution profile, hardness, friability, impurity levels, moisture content, and particle size distribution. In generic drug development, achieving dissolution similarity and bioequivalence with the reference listed drug is particularly important, making dissolution behavior a major CQA. The identification of CQAs enables researchers to focus on parameters that significantly influence product performance and patient outcomes [36-38].
Another major aspect of QbD is the identification and control of Critical Process Parameters (CPPs). CPPs are process variables that have a direct impact on CQAs and therefore require careful monitoring and control during manufacturing. Examples of CPPs include blending speed, granulation time, drying temperature, compression force, coating parameters, and mixing conditions. Variations in these parameters can lead to changes in tablet hardness, dissolution profile, content uniformity, or stability, potentially affecting therapeutic equivalence. By understanding the relationship between CPPs and CQAs, manufacturers can establish robust manufacturing processes capable of consistently producing products that meet quality specifications. Advanced statistical tools such as Design of Experiments (DoE) are widely used in QbD to systematically study the effects of multiple formulation and process variables simultaneously. DoE helps identify critical interactions between variables and optimize formulation composition and manufacturing conditions. This scientific understanding supports the creation of a design space, which is a multidimensional range of material attributes and process parameters demonstrated to assure product quality. Operating within the approved design space provides manufacturers with greater flexibility for process adjustments without requiring extensive regulatory reapproval [40].
Risk assessment is another core component of the QbD framework. Pharmaceutical development involves numerous variables that may influence product quality, and risk assessment tools help identify, analyze, and prioritize potential risks associated with formulation and manufacturing processes. Commonly used risk assessment tools include Failure Mode and Effects Analysis (FMEA), Ishikawa diagrams, Hazard Analysis and Critical Control Points (HACCP), and risk ranking methods. These tools help manufacturers identify high-risk variables that require tighter control and monitoring. Risk assessment also facilitates resource optimization by focusing development efforts on critical factors affecting product quality. Once risks are identified, appropriate control strategies are established to minimize variability and ensure consistent product performance. Control strategies may include in-process monitoring, environmental controls, equipment qualification, raw material specifications, process validation, and real-time analytical testing. Process Analytical Technology (PAT) tools are increasingly integrated into QbD systems to enable continuous monitoring of manufacturing processes and immediate detection of deviations. PAT technologies improve process understanding and support real-time quality assurance rather than relying solely on final product testing [41].
The implementation of QbD in generic drug development offers numerous advantages for pharmaceutical manufacturers and regulatory agencies. QbD improves product quality and consistency by enhancing scientific understanding of formulation and manufacturing processes. It reduces batch failures, manufacturing deviations, and product recalls, thereby lowering production costs and improving operational efficiency. The systematic and knowledge-based approach also facilitates regulatory review because QbD submissions provide detailed scientific justification for formulation and process decisions. Regulatory agencies often encourage QbD-based applications because they demonstrate a proactive commitment to quality assurance and lifecycle management. Furthermore, QbD supports continuous improvement throughout the product lifecycle by enabling manufacturers to make scientifically justified process modifications without compromising product quality. In the context of generic drug development, QbD is especially valuable because it helps ensure consistent bioequivalence and therapeutic equivalence with the innovator product despite potential variability in raw materials or manufacturing conditions.
Despite its advantages, implementation of QbD can present certain challenges, particularly for small and medium-sized pharmaceutical companies. Establishing a comprehensive QbD framework requires significant investment in scientific expertise, advanced analytical instruments, statistical software, and employee training. Extensive experimentation and data analysis are often required during formulation and process optimization, increasing development complexity and initial costs. However, the long-term benefits of improved process robustness, reduced regulatory issues, and enhanced product quality generally outweigh these initial investments. As pharmaceutical industries continue to adopt advanced technologies such as artificial intelligence, machine learning, and automation, QbD is expected to become even more sophisticated and efficient in future pharmaceutical development.
Analytical Method Development and Validation
Analytical method development and validation are essential components of pharmaceutical research, quality control, and regulatory compliance in generic drug development. Analytical methods are used to identify, quantify, and evaluate active pharmaceutical ingredients (APIs), impurities, degradation products, excipients, and finished pharmaceutical products throughout the development and manufacturing process. In generic drug development, analytical methods play a crucial role in ensuring pharmaceutical equivalence, stability, dissolution similarity, and bioequivalence with the reference listed drug. Regulatory agencies such as the United States Food and Drug Administration, European Medicines Agency, and International Council for Harmonisation require scientifically validated analytical methods to guarantee product quality, safety, and consistency. Analytical method development involves selecting suitable analytical techniques and optimizing experimental conditions to achieve accurate, precise, sensitive, and reproducible results. Validation confirms that the developed method consistently performs according to predefined acceptance criteria under specified conditions.
A wide range of analytical techniques are employed in pharmaceutical analysis depending on the nature of the drug substance and formulation. Common analytical methods include spectroscopic, chromatographic, electrophoretic, and hyphenated techniques. Spectroscopic methods such as ultraviolet-visible (UV-Vis) spectroscopy and infrared spectroscopy are frequently used for identification and quantitative estimation of pharmaceutical compounds. Chromatographic techniques such as high-performance liquid chromatography (HPLC), gas chromatography (GC), and thin-layer chromatography (TLC) are widely used for separation, identification, and quantification of APIs and impurities. Advanced hyphenated techniques such as liquid chromatography–mass spectrometry (LC-MS/MS) combine separation and detection capabilities, enabling highly sensitive and selective analysis of complex biological and pharmaceutical samples. The selection of an analytical method depends on several factors including sensitivity requirements, specificity, sample complexity, regulatory expectations, and intended application.
Among the available analytical techniques, HPLC is considered one of the most important and widely used methods in pharmaceutical analysis. HPLC provides excellent precision, sensitivity, reproducibility, and versatility for analyzing APIs, impurities, degradation products, and dissolution samples. In HPLC analysis, compounds are separated based on their interaction with stationary and mobile phases under high pressure. Reverse-phase HPLC is particularly popular in pharmaceutical applications because of its suitability for a broad range of drug molecules. HPLC methods are extensively used for assay determination, content uniformity testing, impurity profiling, dissolution analysis, and stability studies in generic drug development. UV spectroscopy is another commonly used analytical technique due to its simplicity, cost-effectiveness, and rapid analysis capabilities. UV methods are frequently applied for routine assay analysis, dissolution studies, and preliminary formulation evaluation. However, UV methods may have limitations in specificity when complex sample matrices or multiple components are present. LC-MS/MS has become increasingly important in bioanalytical and pharmacokinetic studies because of its exceptional sensitivity and selectivity. This technique is extensively used in bioequivalence studies to quantify drug concentrations in plasma or biological fluids at very low levels. LC-MS/MS combines chromatographic separation with mass spectrometric detection, enabling accurate identification and quantification of analytes even in highly complex biological matrices.
Analytical method validation is a systematic process used to demonstrate that an analytical method is suitable for its intended purpose. Validation parameters are generally established according to ICH Q2 guidelines and include specificity, accuracy, precision, linearity, range, robustness, limit of detection (LOD), limit of quantification (LOQ), and system suitability testing. Specificity refers to the ability of the method to accurately measure the analyte in the presence of impurities, degradation products, or excipients. Accuracy measures the closeness of experimental results to the true value, while precision evaluates reproducibility under repeated testing conditions. Precision is further categorized into repeatability, intermediate precision, and reproducibility. Linearity assesses the ability of the analytical method to produce results directly proportional to analyte concentration within a specified range. Robustness examines the reliability of the method under small deliberate variations in analytical conditions such as pH, flow rate, or temperature. Detection and quantification limits indicate the minimum analyte concentration that can be detected or quantified with acceptable accuracy and precision. System suitability tests are conducted before routine analysis to ensure proper instrument performance and analytical consistency.
Stability-indicating methods are particularly important in pharmaceutical development because they detect changes in drug quality resulting from environmental factors such as temperature, humidity, oxidation, light, and hydrolysis. These methods are designed to accurately separate and quantify the API in the presence of degradation products and impurities. Stability-indicating analytical methods are essential for stability studies conducted according to ICH guidelines, which evaluate the shelf life and storage conditions of pharmaceutical products. Forced degradation studies are commonly performed to intentionally degrade the drug under stress conditions and identify potential degradation pathways. Analytical techniques such as HPLC and LC-MS/MS are extensively used for stability-indicating analysis because of their high sensitivity and separation capabilities. Development of robust stability-indicating methods ensures accurate monitoring of product quality throughout its lifecycle and supports regulatory approval processes.
Overall, analytical method development and validation form the scientific backbone of generic drug development and pharmaceutical quality assurance. Reliable analytical methods ensure accurate characterization of APIs, consistent product quality, successful bioequivalence evaluation, and compliance with global regulatory standards. Continuous advancements in analytical technologies, automation, and computational tools are further enhancing the precision, efficiency, and reliability of pharmaceutical analysis, thereby supporting the development of safe, effective, and high-quality generic medicines [42].
Bioavailability and Bioequivalence Concepts
Bioavailability and bioequivalence are fundamental concepts in pharmaceutical sciences and play a central role in the development, evaluation, and regulatory approval of generic medicines. These concepts are essential for ensuring that a generic product performs therapeutically in the same manner as the innovator or branded product. Bioavailability refers to the rate and extent to which an active pharmaceutical ingredient (API) is absorbed from a dosage form and becomes available at the site of action or in systemic circulation. It is a critical pharmacokinetic property because the therapeutic effectiveness of a drug depends not only on the amount administered but also on how efficiently it reaches systemic circulation. Bioavailability is influenced by several factors including drug solubility, dissolution rate, permeability, gastrointestinal pH, gastric emptying time, first-pass metabolism, formulation characteristics, and route of administration. Drugs administered intravenously are considered to have 100% bioavailability because they directly enter systemic circulation without absorption barriers. However, orally administered drugs often exhibit lower bioavailability due to incomplete absorption and metabolic degradation. Assessment of bioavailability is particularly important during formulation development because even minor variations in formulation composition or manufacturing processes can alter drug absorption and therapeutic performance [40].
Bioequivalence, on the other hand, refers to the absence of a significant difference in the rate and extent of absorption between two pharmaceutical products when administered under similar experimental conditions. In generic drug development, bioequivalence studies compare the test product (generic formulation) with the reference listed drug (RLD) to establish therapeutic equivalence. Regulatory agencies such as the United States Food and Drug Administration, European Medicines Agency, and World Health Organization require generic manufacturers to demonstrate bioequivalence before approval. The primary purpose of bioequivalence testing is to ensure that the generic product delivers the drug into systemic circulation at a similar rate and extent as the innovator product, thereby producing the same therapeutic effect and safety profile. Bioequivalence studies are generally conducted in healthy human volunteers under carefully controlled clinical conditions using randomized crossover study designs. The concentration of the drug in plasma or blood samples is measured over time, and pharmacokinetic parameters are statistically compared between the test and reference products.
The importance of bioavailability and bioequivalence in generic drug approval cannot be overstated because they form the scientific basis for therapeutic interchangeability between generic and branded medicines. Generic manufacturers are not required to repeat extensive clinical efficacy and safety studies performed for innovator drugs because the efficacy of the API has already been established. Instead, demonstrating bioequivalence is considered sufficient evidence that the generic product will perform similarly in patients. This approach significantly reduces the cost and duration of generic drug development, thereby promoting availability of affordable medicines without compromising quality or therapeutic effectiveness. Regulatory approval based on bioequivalence studies also helps avoid unnecessary repetition of human and animal clinical trials, supporting ethical principles in pharmaceutical research. Bioequivalence assessment is especially important for drugs with narrow therapeutic indices, modified-release formulations, and drugs exhibiting high pharmacokinetic variability. In such cases, even small differences in absorption can lead to therapeutic failure or adverse drug reactions, requiring stricter regulatory evaluation and monitoring.
Pharmacokinetic parameters are the primary tools used to evaluate bioavailability and bioequivalence. The most important pharmacokinetic parameters include maximum plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax), and area under the plasma concentration-time curve (AUC). Cmax represents the peak concentration of the drug in systemic circulation and provides information about the rate and extent of absorption. Tmax indicates the time required to reach peak plasma concentration and reflects the absorption rate of the drug. AUC is considered one of the most important parameters because it represents the total systemic exposure to the drug over time and reflects the extent of absorption [41].
Additional pharmacokinetic parameters such as elimination half-life (t½), elimination rate constant (Ke), and mean residence time (MRT) may also be evaluated depending on the study design and drug characteristics. Statistical analysis of bioequivalence data typically involves calculating the 90% confidence intervals for the ratios of Cmax and AUC between the test and reference products. Regulatory agencies generally consider products bioequivalent if these confidence intervals fall within the acceptable range of 80–125%.
The concept of bioavailability and bioequivalence has evolved significantly with advancements in pharmaceutical sciences and regulatory harmonization. Modern analytical technologies such as liquid chromatography–mass spectrometry (LC-MS/MS), population pharmacokinetic modeling, physiologically based pharmacokinetic (PBPK) modeling, and in-silico simulations have improved the accuracy and efficiency of bioequivalence assessment. The Biopharmaceutics Classification System (BCS) has further simplified bioequivalence evaluation for certain highly soluble and highly permeable drugs by allowing biowaivers based on comparative dissolution studies instead of in-vivo clinical studies. Despite these advancements, bioequivalence assessment remains scientifically challenging for complex formulations such as liposomes, inhalation products, transdermal systems, and biologics. Continuous improvements in analytical methods, regulatory guidelines, and computational modeling are helping address these challenges and enhance the reliability of bioequivalence studies. Overall, bioavailability and bioequivalence concepts are indispensable for ensuring the quality, efficacy, safety, and affordability of generic medicines in modern healthcare systems.
Design of Bioequivalence Studies
The design of bioequivalence studies is a critical component of generic drug development because it provides scientific evidence that a generic product is therapeutically equivalent to the innovator or reference listed drug (RLD). Bioequivalence studies are carefully planned clinical investigations conducted under standardized conditions to compare the rate and extent of drug absorption between test and reference formulations. Regulatory authorities such as the United States Food and Drug Administration, European Medicines Agency, and Central Drugs Standard Control Organization have established comprehensive guidelines governing the design, conduct, analysis, and reporting of bioequivalence studies. Proper study design is essential to minimize variability, ensure statistical reliability, protect participant safety, and generate valid pharmacokinetic data for regulatory evaluation. The design of bioequivalence studies includes several important elements such as study protocol development, selection of study design, subject recruitment, inclusion and exclusion criteria, fasting and fed conditions, ethical considerations, and informed consent procedures [40-42].
The study protocol serves as the foundation of a bioequivalence study and provides a detailed plan describing study objectives, methodology, statistical analysis, safety monitoring, and operational procedures. A well-designed protocol ensures standardization and scientific consistency throughout the study. The protocol typically includes details regarding study population, dosage regimen, washout period, blood sampling schedule, analytical methods, pharmacokinetic analysis, and adverse event monitoring. Regulatory agencies require bioequivalence study protocols to comply with Good Clinical Practice (GCP) guidelines to ensure participant safety, data integrity, and ethical conduct. Before initiation, the protocol must be reviewed and approved by an Institutional Ethics Committee or Institutional Review Board. The study protocol also specifies the statistical methods used to compare pharmacokinetic parameters such as Cmax and AUC between the test and reference products. Standardization of study procedures is essential to reduce variability arising from environmental factors, food intake, subject activity, and sample handling conditions.
The most commonly used bioequivalence study design is the randomized two-treatment, two-period, two-sequence crossover design. In this design, each subject receives both the test and reference products in separate study periods with a washout interval between treatments to eliminate residual drug effects. Subjects are randomly assigned to one of two treatment sequences, ensuring balanced distribution and minimizing selection bias. The crossover design offers several advantages because each participant serves as his or her own control, thereby reducing inter-subject variability and improving statistical power. The washout period is generally based on the elimination half-life of the drug and is usually long enough to ensure complete elimination of the previously administered formulation before the next treatment period. In some situations, parallel study designs may be used instead of crossover studies, particularly for drugs with very long half-lives, biologics, or highly variable pharmacokinetic behavior. In a parallel design, separate groups of subjects receive either the test or reference product without crossover. Although parallel designs reduce the risk of carryover effects, they generally require larger sample sizes because of increased inter-subject variability.
Selection of study subjects is another crucial factor affecting the reliability and validity of bioequivalence studies. Most bioequivalence studies are conducted in healthy adult volunteers because healthy subjects generally exhibit lower physiological variability and fewer confounding factors compared to patient populations. Healthy volunteers also reduce the risk of disease-related alterations in drug absorption, metabolism, distribution, or elimination. Regulatory agencies typically recommend inclusion of male and female subjects between 18 and 55 years of age with normal body mass index (BMI) and no clinically significant medical conditions. The sample size of the study is determined based on statistical power calculations, expected variability, and regulatory requirements. Sufficient sample size is necessary to detect meaningful differences between formulations and ensure accurate estimation of pharmacokinetic parameters.
Inclusion and exclusion criteria are established to ensure subject safety and minimize variability in pharmacokinetic outcomes. Inclusion criteria generally include healthy individuals who are non-smokers or light smokers, free from significant medical illnesses, and not taking medications that may interfere with the study drug. Subjects must also have normal laboratory values for hematology, liver function, kidney function, and cardiovascular health. Exclusion criteria typically include history of hypersensitivity to the study drug, gastrointestinal disorders affecting absorption, alcohol or drug abuse, pregnancy, participation in recent clinical studies, and use of medications that may alter drug metabolism. Careful screening of participants helps ensure study consistency and protects volunteer safety throughout the clinical investigation.
Bioequivalence studies may be conducted under fasting or fed conditions depending on the labeling recommendations and absorption characteristics of the drug product. Fasting studies are generally performed after an overnight fast of at least 8–10 hours to minimize variability associated with food intake. In fed bioequivalence studies, subjects consume a standardized high-fat, high-calorie meal before drug administration to evaluate the effect of food on drug absorption. Food can significantly alter gastric emptying time, gastrointestinal pH, bile secretion, and drug solubility, thereby affecting bioavailability. Regulatory agencies often require both fasting and fed studies for orally administered products when food is known to influence drug absorption. Standardized meal composition and timing are essential to ensure consistency across participants and study periods.
Ethical considerations are fundamental in the design and conduct of bioequivalence studies because these studies involve administration of pharmaceutical products to human volunteers. All studies must comply with ethical principles outlined in the Declaration of Helsinki and Good Clinical Practice guidelines. Prior to participation, volunteers must provide written informed consent after receiving detailed information regarding study objectives, procedures, potential risks, benefits, and their rights as research participants. Informed consent ensures that participation is voluntary and based on adequate understanding of the study. Ethical review committees evaluate study protocols to ensure that risks are minimized and participant welfare is protected. Safety monitoring is conducted throughout the study to identify and manage adverse events or unexpected reactions. Confidentiality of participant data must also be maintained according to regulatory and ethical requirements.
Overall, the design of bioequivalence studies is a scientifically rigorous and ethically regulated process aimed at generating reliable pharmacokinetic data for generic drug approval. Proper study design minimizes variability, ensures participant safety, and supports accurate demonstration of therapeutic equivalence between generic and innovator products. Advances in clinical pharmacology, analytical technologies, and statistical modeling continue to improve the precision and efficiency of bioequivalence study design, ultimately contributing to the development of safe, effective, and affordable generic medicines worldwide.
Future Perspectives
The future of generic drug development is expected to undergo remarkable transformation due to continuous advancements in pharmaceutical sciences, regulatory modernization, digital technologies, and increasing global healthcare demands. Generic medicines have already established themselves as essential components of healthcare systems worldwide by improving access to affordable therapies and reducing healthcare expenditure. However, future pharmaceutical landscapes are likely to move beyond conventional generic development toward more patient-centered, technologically advanced, and globally harmonized approaches. One of the most promising future directions is the emergence of personalized generic medicines. Traditional generic formulations are generally developed using a “one-size-fits-all” concept, where standardized doses are intended for broad patient populations. However, advancements in pharmacogenomics, precision medicine, and individualized therapy are gradually shifting the focus toward tailoring drug therapy according to genetic, physiological, and metabolic differences among patients. Personalized generic medicines may involve modified dosage strengths, patient-specific drug delivery systems, or customized formulations designed to optimize therapeutic outcomes while minimizing adverse effects. Technologies such as 3D printing, artificial intelligence-driven dose optimization, and pharmacogenetic screening may enable manufacturers to produce highly individualized generic products in the future. These developments could be particularly beneficial for pediatric, geriatric, oncology, and chronic disease patients who often require personalized dosing regimens.
Regulatory harmonization is another important future perspective in the generic pharmaceutical industry. At present, pharmaceutical companies often face challenges due to variations in regulatory requirements among different countries and regions. Differences in bioequivalence criteria, documentation standards, stability requirements, labeling formats, and manufacturing guidelines can complicate global product development and delay market access. Future efforts are expected to focus on strengthening international collaboration and harmonization of regulatory standards through organizations such as the International Council for Harmonisation and the World Health Organization. Harmonized regulatory systems can reduce duplication of studies, accelerate approval timelines, minimize development costs, and facilitate faster global distribution of generic medicines. Regulatory reliance models and mutual recognition agreements between agencies may also become more common, allowing regulatory authorities to use scientific assessments performed by trusted international agencies. Such initiatives will improve efficiency in drug approval processes and ensure consistent quality standards across international markets. In addition, future regulatory frameworks are likely to incorporate more flexible and science-based approaches such as real-time release testing, continuous manufacturing, and advanced risk-based quality systems.
Emerging technologies are expected to revolutionize every stage of generic drug development, manufacturing, and quality assurance. Artificial intelligence (AI), machine learning, big data analytics, and automation are increasingly being integrated into pharmaceutical research and development processes. AI-driven predictive modeling can help optimize formulations, predict drug-excipient interactions, estimate bioequivalence outcomes, and improve manufacturing efficiency. Machine learning algorithms can analyze large datasets to identify critical process variables, detect quality deviations, and support decision-making during pharmaceutical development. Continuous manufacturing technologies are also gaining attention because they offer improved process efficiency, reduced manufacturing time, enhanced product consistency, and lower production costs compared to traditional batch manufacturing systems. Advanced analytical technologies such as Process Analytical Technology (PAT), real-time monitoring systems, and automated quality control platforms will further strengthen pharmaceutical quality assurance and reduce manufacturing variability. Moreover, nanotechnology, lipid-based drug delivery systems, and advanced modified-release formulations are expected to expand opportunities for development of complex generic products that were previously difficult to replicate. Digital health technologies, electronic clinical trial systems, and blockchain-based supply chain management may also improve transparency, traceability, and regulatory compliance throughout the pharmaceutical product lifecycle.
The global generic pharmaceutical market is expected to continue expanding rapidly due to increasing healthcare needs, patent expirations of blockbuster drugs, and rising demand for cost-effective therapies. Developing countries, in particular, present enormous opportunities for growth because affordable medicines are essential for improving healthcare accessibility in resource-limited settings. Countries such as India, China, and Brazil are emerging as major centers for generic drug manufacturing and export due to their strong pharmaceutical infrastructure, skilled workforce, and competitive production costs. India, often referred to as the “pharmacy of the world,” is expected to play an even greater role in supplying affordable medicines to international markets, particularly in the fields of antiretroviral therapy, oncology drugs, antibiotics, and chronic disease management. In addition, increasing demand for biosimilars and complex generics offers significant opportunities for pharmaceutical companies willing to invest in advanced research and manufacturing capabilities. Patent expirations of several high-value biologics are expected to create major growth potential in the biosimilar sector, which may become one of the fastest-growing segments of the pharmaceutical industry.
Despite these promising developments, the future generic pharmaceutical industry will also face several challenges. Increasing regulatory expectations, intense market competition, pricing pressures, intellectual property disputes, and the technical complexity of modern formulations may create obstacles for manufacturers. Development of complex generics, inhalation products, transdermal systems, and biologics requires sophisticated analytical techniques, advanced manufacturing technologies, and extensive characterization studies. Maintaining data integrity, cybersecurity, and supply chain resilience will also become increasingly important in digitally integrated pharmaceutical environments. Nevertheless, ongoing scientific advancements, international collaboration, and technological innovation are expected to strengthen the overall efficiency, quality, and accessibility of generic medicines globally.
CONCLUSION
Generic drug development and bioequivalence studies play a crucial role in modern healthcare systems by ensuring the availability of safe, effective, and affordable medicines to patients worldwide. Generic medicines have significantly transformed the pharmaceutical industry by reducing treatment costs, improving healthcare accessibility, and promoting competition within the global drug market. The development of generic pharmaceutical products is based on demonstrating pharmaceutical equivalence and bioequivalence with the innovator or reference listed drug, thereby eliminating the need for repetitive and costly clinical efficacy trials. Regulatory authorities such as the United States Food and Drug Administration, European Medicines Agency, Central Drugs Standard Control Organization, and World Health Organization have established stringent guidelines and scientific standards to ensure that generic products meet the same quality, safety, and therapeutic performance requirements as branded medicines.
The process of generic drug development involves several critical stages, including selection of the reference listed drug, preformulation studies, API characterization, formulation optimization, excipient selection, manufacturing process development, analytical method validation, and stability testing. Bioequivalence studies represent the cornerstone of generic drug approval because they scientifically establish that the generic product delivers the drug into systemic circulation at a similar rate and extent as the innovator product. Pharmacokinetic parameters such as Cmax, Tmax, and AUC are extensively used to evaluate therapeutic equivalence between test and reference formulations. Advances in analytical technologies, statistical modeling, and bioanalytical techniques have greatly improved the accuracy and reliability of bioequivalence assessment.
The implementation of Quality by Design (QbD), risk-based quality systems, and advanced manufacturing technologies has further enhanced the robustness and consistency of generic pharmaceutical products. Emerging technologies such as artificial intelligence, machine learning, continuous manufacturing, nanotechnology, and in-silico modeling are expected to revolutionize future pharmaceutical development processes. In addition, global regulatory harmonization and international collaboration are likely to simplify approval pathways and improve worldwide access to high-quality generic medicines. The growing importance of biosimilars, complex generics, and personalized pharmaceutical therapies also presents new opportunities for innovation and market expansion.
Despite significant achievements, the generic pharmaceutical industry continues to face challenges related to regulatory complexity, intellectual property issues, market competition, and development of technically challenging formulations. Nevertheless, the future of generic medicines remains highly promising due to increasing healthcare demands, rising chronic disease burden, and continuous patent expirations of innovator drugs. Generic pharmaceuticals will continue to play a vital role in supporting public health systems, reducing healthcare expenditure, and ensuring equitable access to essential medicines across developed and developing countries. Overall, robust generic drug development practices and scientifically sound bioequivalence studies remain fundamental for maintaining therapeutic confidence, regulatory compliance, and global healthcare sustainability.
REFERENCES

Abhishek Awasthi, Mohit Gupta, Generic Drug Development and Bioequivalence Study, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 369-391. https://doi.org/10.5281/zenodo.20492371
 10.5281/zenodo.20492371
10.5281/zenodo.20492371