
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Quality Assurance, Vidya Niketan Institute of Pharmacy and Research Center Bota, Sangamner Maharashtra, India
Impurity control in oral solid dosage forms represents a convergence of analytical chemistry, formulation science, and regulatory governance with direct patient safety implications, yet the increasing chemical complexity of tablet and capsule matrices continues to challenge conventional quality assurance frameworks. This review addresses the mechanistic, analytical, and regulatory dimensions of impurity profiling in solid oral dosage forms by examining the ICH Q3-series classification architecture and its dose-dependent threshold logic, the specific pathways through which hydrolysis, peroxide-mediated oxidation, Maillard reactions, and excipient-derived nitrite species generate impurities within the constrained solid-state matrix, and the tiered analytical strategy from routine reversed-phase HPLC and UPLC through LC-MS/MS, LC-HRMS, GC-MS, ICP-MS, and NMR that translates regulatory threshold requirements into actionable quality data. The nitrosamine contamination crisis originating with valsartan in 2018 is examined as a paradigm-shifting event that reoriented pharmaceutical quality assurance toward genotoxic risk management in finished products, including the emerging challenge of nitrosamine drug substance-related impurities and the Carcinogenic Potency Categorization Approach adopted by both the FDA and EMA in 2023. The review further demonstrates how Quality by Design principles, embedded from the Quality Target Product Profile through risk assessment, design space definition, and Process Analytical Technology surveillance, transform impurity control from reactive end-product testing into predictive manufacturing governance of critical quality.
The safety profile for any pharmaceutical product is largely influenced by the pharmacological properties of the active ingredient, but also strongly influenced by the nature, identity and levels of other chemical entities that may exist with it, termed impurities [1]. Any substance in the drug product which is not the drug substance (also called the drug product component) nor any of the excipients used in the manufacture of the drug product is an impurity as defined in ICH Q6A and even low levels of these impurities may have consequences from subtle decreases in potency, to serious toxicological effects in patients [2]. Managing the consideration of impurities has become a key part of product quality design and this has been reflected in successive iterations of guidance from the regulatory community, which now covers every phase of a drug product lifecycle [3]. Oral solid dosage forms such as tablets, capsules and their film-coated counterparts are the most prevalent form of pharmaceutical therapy globally, and they are particularly complex with respect to impurities [4]. In contrast, when the composition is in the liquid state, the mobility of molecules leads to reasonable prediction of the rate of degradation in the solution phase, whereas in solid dosage forms there is a complex and constrained matrix in which water activity and excipient chemistry is coupled to mechanical stresses and environmental conditions, and their resulting interactions can be predicted and monitored with difficulty [5]. Most reactions which can lead to the formation of impurities in tabletted or capsuled material involve small molecules (e.g. water, hydrolytic agent; aldehydes and peroxides arising from the excipients, oxidative initiators; metal ions as catalysts) and may proceed without any noticeable changes of the product's physical appearance during storage [6].
The guidelines and standards for impurities in solid oral dosage forms have been significantly changed. The ICH Q3 series (Q3A for drug substance, Q3B for drug product, Q3C for residual solvents, Q3D for elemental impurities) laid the groundwork for the classification and threshold system [7]. The subsequent development of ICH M7(R2), dealing with mutagenic and genotoxic impurities including nitrosamines (which became an important category of impurities due to significant concerns raised in the literature), introduced a change in approach to the assessment of impurities from mass-based limits to acceptable daily intake limits based on cancer risk [8]. Impurity science was recently illustrated to be anticipating more than only typical degradation pathways when N-nitrosodimethylamine was found in valsartan-containing tablets in 2018, which led to global recalls and a complete re-evaluation of the potential for nitrosamine impact in various drug classes in real world storage conditions. In this context, the significance of advanced analytical methodology has emerged as the focus in the pharmaceutical quality assurance [9]. The shift from HPLC methods that could only handle the detection of known degradation products to high resolution mass spectrometry platforms that can handle sub ppm detection of unknowns has been driven by both scientific and regulatory needs. At the same time, the introduction of the Quality by Design (QbD) approach under ICH Q8 (R2) and ICH Q9 (R1) has transformed impurity control from a testing challenge to an engineering challenge, which requires close collaboration between analytical scientists and formulation scientists throughout drug development [10].
This review covers classification of impurities in oral solid dosage products, detection, regulatory challenges arising from nitrosamines, QbD-based approaches to impurity control, validated methods and the technologies which will be key in the next decade of the field. The discussion is organised to benefit pharmaceutical scientists, analytical chemists and regulatory bodies involved in the development and quality control of solid oral pharmaceutical products.
2. Classification of Impurities in Oral Solid Dosage Forms: Origins, Types, and Regulatory Definitions
Pharmaceutical impurities are not a single entity but rather a complex mixture of chemically complex impurities of diverse origin, structural similarity or dissimilarity to the active pharmaceutical ingredient, and differing toxicological properties [11]. Whereas a rigorous classification framework cannot be just an academic abstraction, it is rather the operational prerequisite that dictates the analytical strategy to be employed, the threshold to be used for the regulatory assessment and whether or not safety qualification data need to be generated prior to the product being allowed to be marketed [12]. The ICH Q3 definition of organic impurities in drug substances includes by-products of the synthesis process, degradation products from the drug substance, and the by-products of the starting materials used in the synthesis of the product, which may occur during the synthesis process or during storage [13]. The main organic impurity issue for drug products regulated by ICH Q3B(R2) is no longer related to process-related impurities, but to degradation products that might occur as a result of API-excipient, API-water, API-oxygen, and API-light reactions during the shelf life of the product [14]. The difference has scientific significance, because if the API released from manufacturing is contaminated with impurities and after 24 months of storage at accelerated conditions, the impurity profile of the API becomes different from the API in the finished tablet, and the regulatory specification must consider both [14]. Inorganic impurities, such as residual catalysts, heavy metals and inorganic salts from the reagents, originate predominantly from the synthetic route and are managed by ICH Q3A(R2) for the drug substance and ICH Q3D(R2) that provides a risk-based approach for elemental impurities with permitted daily exposure values for 24 elements grouped according to safety concern [15].
Residual solvents are a subset of physically unique impurities: an organic chemical that is a volatile residue in the final drug product due to its use in the API synthesis or formulation process. ICH Q3C(R8) categorizes them as Class 1 solvents (to be avoided completely or limited to very low levels, e.g., benzene, carbon tetrachloride); as Class 2 solvents (have non-negligible toxicity and must be controlled based on permitted daily exposure, e.g., acetonitrile [410 ppm], methanol [3000 ppm], toluene [890 ppm]); and as Class 3 solvents (have low toxicity and are acceptable without specific toxicological qualification, e.g., ethanol, acetone, [5000 ppm]) [16]. The concept of genotoxic and mutagenic impurities as a separate regulatory class, with a limit under ICH M7 (R2), was a shift from the mass-fraction threshold approach used for conventional organic impurities [17]. The acceptable daily intake (ADI) limits for genotoxic species are based on a linear extrapolation cancer risk model that seeks to limit lifetime excess cancer risk to 1 in 100,000 and concentrations that are much lower than those that elicit conventional toxicological endpoints. This means that a threshold of toxicological concern (TTC) of 1.5 micrograms per day, assuming lifelong exposure, is reached for a generic genotoxic impurity, which is far lower than the qualification thresholds of 0.15% or 1 mg per day intake for conventional organic impurities in accordance with the Q3A and Q3B guidelines [18]. Nitrosamine impurities are now considered to fall into the genotoxic category and have established acceptable daily intakes (ADIs) set separately by the regulatory agencies, with NDMA listed at a maximum daily intake of 96 nanograms per day in most jurisdictions [19].
One of the practical importance is the difference between the impurity profiles of drug substances and drug products, especially for oral solid dosage forms. The pharmacopoeial monographs of drug substances focus on the process-related impurities that are generated during the synthetic process, and the monograms for dosage forms focus on impurities that are generated during storage [20]. Impurities from excipients such as reactive aldehydes from lactose, polyethylene glycol (PEG) or polyvidone (PVP) impurities, including peroxides, and residual formic acid from some cellulosic polymers are therefore not found in API specifications, but represent an additional impurity reservoir unique to the drug product matrix. The various categories of impurities in oral solids and the regulatory guidance, thresholds, and other conditions that apply to each are tabulated below [21].
TABLE 1: Classification of Pharmaceutical Impurities in Oral Solid Dosage Forms Types, Origins, Applicable ICH Guidelines and Reporting/Identification/Qualification Thresholds [22,23].
|
Impurity Category |
Primary Origin in Solid Dosage Form |
Applicable ICH Guideline |
Reporting Threshold |
Identification Threshold |
Qualification Threshold |
|
Organic process impurities |
Synthetic intermediates, reagents, by-products in API synthesis |
Q3A(R2) |
0.03% |
0.10% (≤2 g MDD); 0.05% (>2 g MDD) |
0.15% or 1.0 mg/day (lower applies) |
|
Degradation products |
API–excipient interactions, hydrolysis, oxidation, photodegradation during storage |
Q3B(R2) |
0.05% (≤1 g MDD); 0.03% (>1 g MDD) |
0.10% or 1 mg/day |
0.15% or 1 mg/day (lower applies) |
|
Residual solvents |
Manufacturing/ processing solvents retained in API or excipients |
Q3C(R8) |
Based on PDE |
PDE-based class limit |
Class 1: avoid; Class 2: PDE-defined; Class 3: ≤5000 ppm |
|
Elemental impurities |
Catalysts, raw materials, equipment, excipients |
Q3D(R2) |
Risk-based |
Based on PDE per route of exposure |
Component or product option testing; ICP-MS/ICP-OES required |
|
Genotoxic/ mutagenic impurities |
Synthetic route by-products, excipient reactions, nitrosamine precursors |
ICH M7(R2) |
Below TTC |
Structural alert assessment |
TTC: 1.5 µg/day (lifetime exposure); individual AI limits for nitrosamines (e.g., NDMA: 96 ng/day) |
|
Excipient-derived reactive impurities |
Peroxides (PEG, PVP), aldehydes (lactose, starch), formic acid (HPC, HPMC) |
Q3A/Q3B contextual |
Case-specific |
Case-specific |
Based on reaction product identity and ICH M7 if genotoxic |
|
Chiral/ enantiomeric impurities |
Asymmetric synthesis, racemization under stress conditions |
Q3A(R2)/Q6A |
0.03% |
As per Q3A |
Biological activity-based determination |
3. Sources of Impurity Formation Specific to Oral Solid Dosage Forms
The molecular environment within an oral solid dosage form is constrained and therefore impurity-generating conditions and opportunities are different from those found in solution-based pharmaceutical systems. Molecular diffusion in the solid state is so restricted that reactive species created inside the API crystal lattice or at the API/excipient interface will have to interact via relatively few pathways [24]. This physical limitation is not a limitation in terms of reactivity of small electrophilic and oxidative species, and the total exposure over the product's shelf life (which can range from 24 to 36 months under real-time storage conditions) is enough to cause significant chemical changes. The specific mechanisms by which impurities are generated in tablets and capsules depends not only on the intrinsic chemical susceptibilities of the API, but also on the chemistry of the excipients themselves that is capable of generating the impurities [25]. Hydrolysis is the most common degradation mechanism in moisture sensitive functional groups like ester, amides, lactams, and glycosidic linkage in solid dosage forms. The water activity of a tablet is a significant difference from solution, but even tightly controlled solid dosage forms will absorb water from the atmosphere, and the water that is absorbed onto the surface of excipients can catalyze hydrolytic reactions at the API/excipient interface [26]. The content of hydrolysis is directly proportional to the moisture vapor transmission rate of the packaging system and the hygroscopic properties of individual excipients as well as the moisture content in the formulation at equilibrium. Hydrolysis to salicylic acid in commercial tablet formulations of aspirin is one of the earliest and most widely investigated moisture-catalysed hydrolytic degradation of oral solid formulations [27].
In solid oral dosage, oxidative degradation can occur by two major pathways: autoxidation, which occurs in presence of molecular oxygen that enters the dosage form through packaging defects or head space; peroxide-mediated oxidation, which involves the presence of reactive oxygen species generated from excipients [28]. In many commercial tablet formulations, the latter is quantitatively more important. Trace hydroperoxide impurities are found in polymeric excipients resulting from free radical autoxidation of carbon–hydrogen bonds in the vicinity of heteroatoms during the manufacturing processes of these excipients: povidone (PVP K-30), crospovidone, polyethylene glycol grades (PEG 300, 400, 600), polyethylene oxide, hydroxypropyl cellulose, and polysorbate 80. For example, the peroxide content of povidone has been found to rise step-by-step with storage at higher temperatures for the drum-dried and belt-dried grades [29]. Hartauer and co-workers then conclusively showed that the N-oxide degradant of raloxifene, which causes oxidative conversion in tablet formulations, is due to the presence of peroxide impurities in povidone and crospovidone, leading to a rational peroxide limit test for these excipients as a process control strategy. The Maillard reaction is another type of impurity and is a common pathway in tablet formulations of primary or secondary amine APIs and reducing sugar excipients [30]. A condensation reaction between the amine group and the carbonyl function of the reducing sugar (typically lactose monohydrate, but also glucose as a reducing sugar impurity in the microcrystalline cellulose) forms a Schiff base intermediate which undergoes Amadori rearrangement to ultimately produce brown coloured melanoidin products and furanose derivatives such as 5-hydroxymethylfurfural. The mechanisms of the Maillard reaction between lactose and fluoxetine hydrochloride (a secondary amine) and between vigabatrin (a primary amine drug) and the monosaccharide glucose, which was found as a trace impurity in microcrystalline cellulose, have been well characterised. In the same way, amlodipine besylate in solid dosage form experiences glycosyl-mediated Maillard reaction to form a glycosyl adduct, which can be identified by LC-MS [31].
Another source of impurities in solid oral forms is the formation of formaldehyde and formic acid during processing or storage that results from the oxidative degradation of polyethylene glycols and polysorbates, which is not as widely discussed, but is analytically significant in this context. The degradation of BMS-204352 in a solid formulation was determined to be due to formaldehyde released from PEG 300. N-methylation and N-formylation of the secondary amine varenicline were both thought to be caused by the presence of formaldehyde released from polyethylene glycol in an osmotic tablet system. Other types of impurities, such as nitrate or nitrite, can be found in some excipients and cause a type of risk not addressed by well-established degradative pathways: they can react with the amine groups of the API under the mildly acidic and aqueous microenvironmental conditions found within tablet formulations to form genotoxic N-nitroso species and are now a focal point for regulatory investigation around oral solid dosage formulations [32].
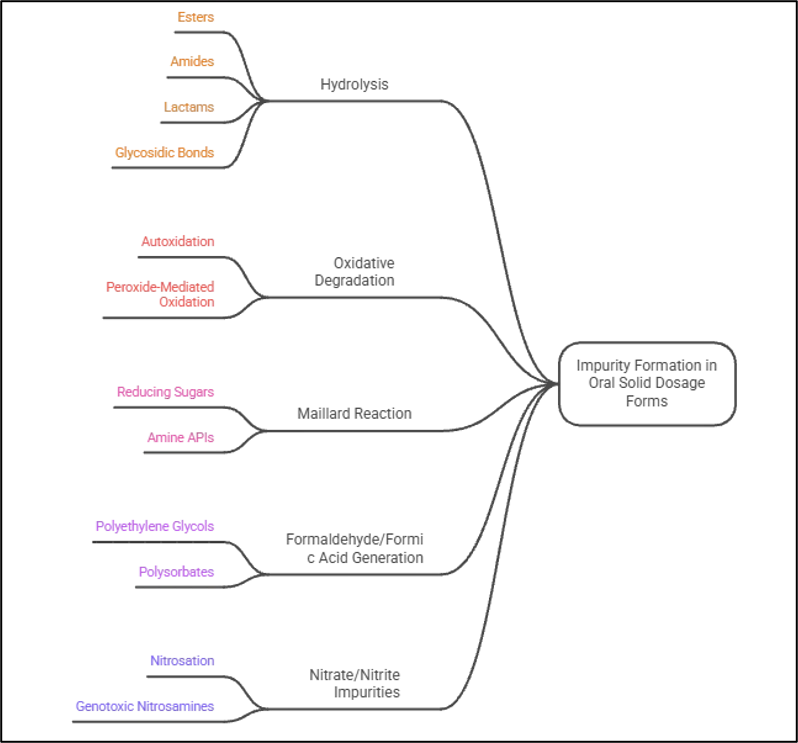
Figure 1. Impurity Formation in Oral Solid Dosage Forms.
4. Analytical Strategies for Impurity Detection and Quantification
The translation of regulatory impurity threshold requirements into actionable regulatory quality control data relies solely on the availability of analytical methods that are capable of reliably detecting, quantifying, and (if required) structurally characterizing the impurities at the concentrations specified in the ICH guidelines [33]. None of the available techniques meet all of these requirements and deliberate selection and integration of complementary analytical platforms calibrated according to the nature of the impurities of interest are required for the practical development of a fit-for-purpose impurity profiling strategy for an oral solid dosage form. For the past 30 years, reversed-phase HPLC with ultraviolet or photodiode array detection has been the standard analytical test for routine impurity profiling in pharmaceutical industries and the regulatory requirement for related substance testing in the NDA and ANDA submissions is established. The widespread applicability of the technique is partly due to the fact that the technique can be performed in a manner that is accessible to operations, compliant with ICH Q2(R2) validation guidelines, and capable of resolving and quantifying impurities down to 0.03% relative to the API (ICH Q3B(R2) reporting threshold) with rigorous method development [34]. The basic problem with the UV based detection method is that it relies on the relative absorptivity of the API and the degradants at the detection wavelength to accurately estimate the API's concentration in the sample when the API concentration is unknown, and that relative absorptivity of API and its degradants is only known if authenticated degradant is prepared and injected along with the sample, which is not available early in development [35].
Ultra performance liquid chromatography (UPLC) uses stationary phase particles of only 1.0 micron or less and pressures up to 100MPa, providing significantly greater resolution, speed and sensitivity than standard HPLC, and achieving an analysis time of about 3 times faster, with a corresponding reduction in peak widths, hence increasing the resolution of the peaks and the signal-to-noise ratio at low levels of impurity. The benefits make UPLC especially useful for gradient based stability indicating methods where separation of structurally similar degradants from the API is essential [36]. A key part of UPLC's performance gain is the direct relationship between the performance increase and the decrease in particle size. Smaller particle size results in smaller eddy-diffusion and mass-transfer terms, which enables operation of faster linear velocity without a decrease in chromatographic efficiency. If the identification of the impurity is required (as per ICH Q3), chromatographic separation alone is not sufficient and mass spectrometric detection is required, which is known as hyphenation [37]. The method is highly selective and sensitive, enabling the accepted limits of intake of genotoxic impurities as defined in ICH M7(R2) to be met, with limits of quantification in the sub-nanogram per millilitre range required for compliance with the limits set by the ICH. To address the structural elucidation of unknown degradants, high resolution mass spectrometry platforms such as Orbitrap and quadrupole time-of-flight devices are able to provide exact molecular formulae with mass accuracy less than 5 ppm, providing the elemental composition data from which structural hypotheses can be constructed. UPLC-HRMS has been established as the primary line of defense to identify unknown impurities that are identified in a forced degradation study of new drug substances and products for solid dosage forms [38].
NMR spectroscopy is a unique and invaluable method for the definitive assignment of the structure of isolated or enriched impurities. Though NMR is not sensitive enough for use in the direct detection of trace amounts of degradation products in complex dosage-form matrices, the ability to provide unambiguous connectivity, stereochemical, and conformational information is invaluable for the characterization of isolated degradants for which identity cannot be confidently assigned using only mass spectrometric data [39]. Moreover, Quantitative NMR has proven itself as a useful method to measure relative response factors when using mixtures containing no pure reference standards, due to the universal and quantitative nature of the proton signal. Residual solvent profiling is a validated method of choice (ICH Q3C) in which headspace GC-MS is used for the analysis of the volatile residues found in Class 1 and Class 2 solvent limits in the low ppm concentration range. Inductively coupled plasma mass spectrometry is capable of the multielement sensitivity and dynamic range required for the elemental impurities determination required by ICH Q3D(R2) that specifies the determination of 24 elements, the limits for which vary several orders of magnitude depending on the element and route of administration [40]. One of the most often overlooked sources for impurity profiling error in solid dosage forms is sample preparation. Co-extraction of excipients can have a suppression or enhancement effect on ionization by LC-MS methods, and extraction efficiency for API and impurities from tablet matrices can vary significantly based on the solvent system, time for tablet to disintegrate and the type of physical processing performed on the sample [41].
TABLE 2: Comparative Overview of Analytical Techniques for Pharmaceutical Impurity Profiling in Oral Solid Dosage Forms [42,43].
|
Technique |
Detection Principle |
Approximate LOQ Range |
Structural Information |
Regulatory Use Context |
Primary Limitations in Solid Dosage Form Matrix |
|
RP-HPLC/ UV-PDA |
UV absorbance at fixed or diode-array wavelengths |
0.01–0.05% w/w of API |
None (retention time only) |
Routine related substance testing; ICH Q2(R2) validation |
Response factor uncertainty without standards; co-elution risk |
|
UPLC/ UV-PDA |
Same as RP-HPLC; sub-2-µm particle column |
0.003–0.03% w/w of API |
None |
High-throughput stability testing; improved gradient resolution |
High system backpressure; more demanding system qualification |
|
LC-MS/ MS (QqQ) |
Multiple reaction monitoring of product ions |
0.1–1 ng/mL (solution) |
Fragment ions; targeted known structures |
Targeted genotoxic impurity quantification; ICH M7 compliance |
Ion suppression from excipient matrix; requires authentic standard |
|
LC-HRMS (Orbitrap, QTOF) |
Exact mass measurement (<5 ppm accuracy) |
0.001–0.01% w/w of API |
Elemental composition, fragmentation patterns |
Unknown degradant elucidation; non-targeted screening |
Expensive instrumentation; requires skilled spectral interpretation |
|
GC-MS |
Electron ionization fragmentation of volatile species |
Low ppm range |
Spectral library matching |
Residual solvent analysis (ICH Q3C); volatile impurities |
Applicable only to volatile/semi-volatile compounds |
|
ICP-MS/ ICP-OES |
Plasma ionization; elemental mass detection |
ppb to sub-ppb range |
Element identity only |
Elemental impurity profiling (ICH Q3D); 24 regulated elements |
Sample preparation (digestion); no organic structural information |
|
NMR (1H, 13C, 2D) |
Nuclear spin relaxation in magnetic field |
>1–10 µg (isolated) |
Complete connectivity and stereochemistry |
Definitive structural confirmation of isolated impurities |
Low sensitivity; requires isolated or enriched impurity samples |
|
Headspace GC-MS |
Volatilization; electron ionization; mass detection |
ppm range in headspace |
Spectral library matching for volatiles |
Residual solvent; nitrosamine precursor analysis |
Limited to volatile species; matrix partition effects |
5. The Nitrosamine Crisis: Genotoxic Impurities, Regulatory Response, and Current Control Strategies
The detection of N-nitrosodimethylamine (NDMA) contamination in valsartan drug substance in mid-2018 is perhaps one of the most significant and immediate regulatory consequences to be observed in the pharmaceutical industry's history of quality assurance [44]. The discovery was made by the manufacturer Novartis during routine testing and was made the basis for a referral under Article 31 of Directive 2001/83/EC under which the European Medicines Agency (EMA) has launched a series of investigations which have uncovered the presence of nitrosamine in the various sartan drug classes (irbesartan, losartan, candysartan, olmesartan) and led to the voluntary withdrawal from the global market of these drugs [45]. By 2019, NDMA had also been found in drug products containing ranitidine, nizatidine and metformin, in a way that was unrelated to the API manufacturing process, thus demonstrating that the NDMA issue was not just a problem with a single synthetic route but a chemically systemic one, based on various formation pathways [46]. The source of NDMA in the original valsartan case was identified, and the source of the nitrite was determined to be for quenching unreacted azide reagents under acidic and thermal conditions with dimethylformamide as a process solvent. Under the hydrolysis condition, dimethylformamide can break down to form dimethylamine, which can be nitrosated with nitrous acid formed in the sodium nitrite quench step to produce NDMA. This mechanism called "secondary amine nitrosation by an in situ nitrosating agent" emerged as a risk factor that was common to all synthetic and formulation pathways where secondary amines and nitrite-generating reagents were found together under acidic aqueous conditions, and led to the introduction of a requirement for manufacturers to review all synthetic and formulation routes for the potential presence of nitrosamines and report the findings to health authorities as a three-step confirmatory review process [47].
The ICH M7(R2) acceptable intake levels for small molecule nitrosamines are based on their classification as a "cohort of concern," which are substances that are likely human carcinogens in various animal species. There are some variations in the AI levels for individual small-molecule nitrosamines, as follows: NDMA is controlled at 96 nanograms per day, NDEA at 26.5 nanograms per day, NMBA at 96 nanograms per day and NDBA at 26.5 nanograms per day in major regulatory jurisdictions [48]. The limits are significantly lower than for traditional "conventional" organic impurities and have forced the development of very sensitive analytical methods that function at levels well below that of typical UV HPLC systems. The FDA has published eight LC-HRMS methods for drug quantification that include nitrosamines, and four LC-HRMS methods validated in USP General Chapter <1469> for six of the major small molecule nitrosamines (NDMA, NDEA, NMBA, NMPA, DIPNA, and NDBA) [49].
A more recent and scientifically more complex aspect of the nitrosamine problem is the nitrosamine drug substance-related impurity (NPS), which is a chemical species that is structurally different from the generic small molecule nitrosamines, but contains the API or an API fragment. While API synthesis does not involve the formation of NDSRIs, they are formed in the final product resulting from a reaction between amine containing API molecules and trace nitrite ions from excipients. It has been estimated that the excipient contribution to the formulation of a typical oral tablet is around 1 microgram per gram of nitrite, which is enough to nitrosate secondary/tertiary amine groups on susceptible APIs in the slightly acidic, aqueous microenvironmental conditions of a tablet. Voluntary recalled products for NDSRI formation include varenicline, propranolol, quinapril and dabigatran etexilate [50]. Even more complex is the case of secondary amine degradants, which are generated from parent APIs that do not contain these secondary amines; for instance, this is the case for metformin and sitagliptin. The acceptance limits for NDSRIs are far more complex than for generic small molecule nitrosamines due to the lack of compound-specific toxicology information. The Carcinogenic Potency Categorization Approach which was adopted by both the FDA and EMA in 2023 fills this gap by using a structure-activity relationship approach that assigns NDSRIs to the four categories of potency, ranging from 1500 nanograms per day (ng/d) at low potency to 27 nanograms per day (ng/d) at high potency, depending on the nature of the alpha-substitution next to the nitroso group, the presence of electron withdrawing or activating substituents, and molecular features associated with metabolic bioactivation. If the classification is ambiguous, the FDA suggests performing an enhanced Ames bacterial reverse mutation test that is performed under conditions that maximize sensitivity toward structurally complex nitrosamines [51].
Nitrosamine risk management is now an essential part of the solid dosage form quality assurance process as it forces drug product formulators to consider the level of nitrite in excipients, whether during product compatibility testing or during the formulation of new products; and to consider mitigation strategies for the API, such as excipient grade selection, pH optimization, excipients containing antioxidants, or reformulation to remove excipient-API combinations that are known to cause nitrosation. The development of the nitrosamine regulatory framework, from the initial valsartan recall in 2018, to the implementation of CPCA in 2023, and the continued FDA deadline extensions for NDSRI confirmatory testing until 2025, have mirrored a quality assurance discipline that is evolving in direct response to the new chemistry risk mechanisms found in finished oral dosage form products [52].
6. ICH-Guided Qualification, Specification Setting and Risk-Based Control Strategies
More than analytical data is needed to translate the detection of an impurity at a specified level and above, into a valid regulatory specification; the required data must be accompanied by a structured, scientifically well-founded qualification process, which is based on the identity of the impurity, and its level of exposure, and then on evidence of biological safety. The ICH Q3 series outlines the qualification process as the systematic gathering of data that an impurity of a specified concentration does not present an unacceptable risk to the patient, with the process being triggered hierarchically according to concentration: an impurity is first reported, then identified if it is found at a concentration above the threshold for identification, and qualified based on safety data if the concentration is above the threshold for qualification [53]. These thresholds are dose dependent and the primary breakpoint for two grams/day is that the qualification threshold is reduced to 0.05%. For conventional organic impurities, the qualification limits as specified in Q3A and Q3B are only applicable to non-mutagenic species and do not apply to impurities that have been shown to contain a structural alert that confirms their potential to be a mutagens or have a positive Ames result, regardless of absolute concentration, as those impurities shall be assessed and controlled based on the ICH M7(R2) TTC-based AI framework [54].
Conventional organic impurities are usually assessed for safety using a comparative nonclinical toxicological evaluation of the drug substance containing the impurity at the proposed specificity level with previously qualified material and/or appropriate literature precedent data demonstrating absence of concern for the level of exposure expected for the impurity. The qualification pathway for NDAs and ANDAs is not identical: while for innovator products, the sponsor will have to generate a toxicological profile for the compound, for ANDAs in many cases, the applicant may rely on the safety information previously generated for the impurity profile of the reference listed drug when the levels of impurities encountered in the generic product are not greater than in the qualified impurity profile of the innovator [55]. Manufacturing changes made after approval which introduce new impurities or raise existing impurities above previously qualified levels will be subject to a fresh qualification evaluation and may require a prior approval supplement or changes being effected filing under the provisions of ICH Q12 change management [56]. The specification-setting process under ICH Q3B(R2) for finished drug products involves the integration of data from development batches, stability batches and clinical study batches and the impurity profile from commercial manufacturing forms the basis for proposed acceptance criteria. One of the core tenets of FDA guidance and academic research of impurity specification practice over the last several years is that batch data experience is important but it is not sufficient to establish the acceptance criteria for a specification the specification needs to be clinically and toxicologically supported and patient-centric whenever scientifically possible and should not simply be based on the range of observed impurity levels in the batches in production. This is especially significant for pharmaceutical development programs where the process development batches could have impurity levels significantly lower than the process could reliably produce at commercial scale, resulting in specification levels that are too tight [57].
The use of Quality by Design concepts including the explicit inclusion of the impurity content as a Critical Quality Attribute in the Quality Target Product Profile as described in the ICH Q8(R2) document has changed the approach to designing impurity control strategies. If impurity formation is identified as a CQA during development, tools for risk assessment such as Ishikawa diagrams and Failure Mode and Effects Analysis are used to identify the critical process parameters (CPPs) and critical material attributes (CMAs) that affect impurity levels, including excipient grade, drying temperature, compression force, film coating conditions, etc., and Design of Experiments (DOE) is used to quantify these and define ranges of operation for the process within which the impurity CQA is within specification. The resulting control strategy, which is described in the regulatory submission, not only includes the final product impurity specification, but also the in-process controls, starting material specifications, and excipient quality specifications that collectively assure compliance. This all-encompassing risk-integrated control framework is far more powerful than just a simple end product test limit, and regulatory agencies have made increasing signals of preference in NDA/ANDA submissions. The current ICH qualification thresholds for the maximum daily dose range are summarized in Table 3 below [58].
TABLE 3: ICH Q3A/Q3B Reporting, Identification, and Qualification Thresholds Across Maximum Daily Dose Ranges with Application Notes for Oral Solid Dosage Forms [59,60]
|
Maximum Daily Dose (MDD) |
Reporting Threshold |
Identification Threshold |
Qualification Threshold |
Special Conditions |
|
Any MDD (drug substance; Q3A) |
0.03% |
0.10% |
0.15% or 1.0 mg/day (lower applies) |
Unusually toxic/ mutagenic impurities: controlled irrespective of level |
|
≤1 g/day (drug product; Q3B) |
0.05% |
0.10% or 1.0 mg/day |
0.15% or 1.0 mg/day (lower applies) |
Unidentified impurities reported as "unidentified A" with RRT |
|
>1 g/day (drug product; Q3B) |
0.03% |
0.05% or 20 mg/day |
0.05% or 20 mg/day (lower applies) |
Higher MDD products require lower thresholds due to greater absolute exposure |
|
Any MDD — genotoxic (ICH M7) |
Below TTC |
Structural alert assessment mandatory |
TTC: 1.5 µg/day (lifetime); AI set individually per compound |
Cohort of concern (N-nitrosamines, aflatoxins): below TTC always |
|
Cancer therapy (ICH S9 exemption) |
Per Q3 A/B but modified |
Per Q3A/B |
Higher limits permissible |
ICH M7 TTC approach not recommended; use individual risk-benefit assessment |
|
ANDA — generics (FDA add-on) |
Same as Q3A/ Q3B |
Justified against RLD impurity profile |
Must not exceed qualified levels for RLD |
Changes above qualified RLD levels require safety data or FDA consultation |
MDD = Maximum Daily Dose; RRT = Relative Retention Time; TTC = Threshold of Toxicological Concern; AI = Acceptable Intake; RLD = Reference Listed Drug; Q3A/B = ICH Q3A(R2)/Q3B(R2)
Figure 2: ICH-Guided Impurity Control
7. Quality by Design as a Proactive Framework for Impurity Control in Solid Dosage Manufacturing
Although the ICH Q3 qualification and specification-setting framework is rigorous, it is fundamentally classical in its application – rather than impurities being detected in the batch, the threshold is met which triggers identification and qualification activity and specification is set based on the level that is present. Quality by Design, based on ICH Q8(R2), Q9(R1) and Q10, represents an epistemological shift of impurity control from end-point testing to the control of system design to provide the end-product with the required quality. The QbD approach to the control of impurities starts by adding and defining the content of the impurities as a Critical Quality Attribute in the Quality Target Product Profile. An impurity content is a physical, chemical, biological or microbiological property that should be within an appropriate limit, range or distribution to ensure desired product quality, and hence the exceedance of this property is directly related to patient safety, so the definition of a CQA can be considered unambiguous for this property. After embedding the impurity content in QTPP as a CQA, the risk assessment phase uses Ishikawa fishbone diagrams to qualitatively identify sources of impurity generation throughout the product's formulation and manufacturing process (from API chemical characteristics to active, environmental conditions, and process steps) and FMEA to quantitatively score the severity, probability of occurrence, and detectability of each of said sources to determine the relative contribution of each of said sources to risk. Those variables with high RPN are then selected for experimental studies [61].
Design of Experiments is the critical experimental element in the impurity control workflow of QbD. Factorial and response surface designs (such as central composite design and Box–Behnken designs) are used to assess the effects of the identified CPPs and critical material attributes on the outcomes of the impurities and the models are then used to create the multidimensional design space within which the process can be operated with confidence that the impurities will maintain within the specification [62]. In general, the following unit operations in the manufacturing process are points of concern for impurity-related CPPs for tablet formulation: granulating fluid volume, concentration of binder, inlet temperature of drying air, outlet temperature of drying air, drying time, residual moisture content, compaction force, dwell time, inlet air temperature, coating solution concentration, and spray rate of the coating solution. The drying step is especially significant for APIs that are hydrolysis sensitive, as any moisture content above a certain point is directly linked to the degradation during the downstream processing and storage. Coating of the tablet with film presents an extra risk of impurities due to thermal exposure of the tablet core to aqueous coating suspensions at high temperature and/or due to the potential introduction of impurity-containing reactive species in coating polymers and plasticizers [63].
The QbD development program yielded a multi-level control strategy: upstream controls at API/excipient level (e.g., peroxide limit testing of PVP and crospovidone, nitrite limit testing of key excipients in amine-containing drug products, and reducing sugar testing of lactose in amine-API formulations); in-process controls during manufacturing (e.g., moisture content of dried granules, and tablet core temperature during coating); and downstream controls in the form of release and stability testing with validated stability-indicating methods. This architecture is significantly better than a one-tier end-product test, since it is tackling the root of impurity formation, instead of just catching it afterwards. Process Analytical Technology (PAT) tools are becoming more well-suited to complement impurity control in QbD by allowing for the ability to monitor the process as you work [64]. The moisture content of granules can be measured in-line during drying by applying near-infrared spectroscopy technology, which can be used to dynamically adjust process parameters to avoid moisture levels responsible for hydrolytic degradation. Raman spectroscopy has been shown to be useful for in-line monitoring of polymorphic form during granulation and milling, where polymorphic transitions under the effects of mechanical stresses may lead to the production of structurally different impurities. When used within a QbD context, these PAT tools provide the basis for real-time release testing as called for by ICH Q10, as opposed to fixed interval end-product impurity testing, which follows a continuous quality surveillance model [64].
8. Future Perspectives
The landscape of impurity profiling and control in oral solid dosage forms is on the verge of undergoing significant change, fueled by three key trends: the scientific challenges of NDSRI risk assessment remain unresolved, computational and artificial intelligence technologies are maturing to become more predictive in impurity science, and regulatory agencies and the pharmaceutical industry are pressuring analytical quality assurance to become more sustainable, quicker and more predictive. The incorporation of artificial intelligence and machine learning in the application of QSAR models already started to change the face of the ICH M7(R2). The current regulatory framework approved two-model QSAR approaches, which incorporate such expert rule-based systems and statistical models, as first tier screening tests for structural mutagenicity alerts, and the false-negative rates in the combined analysis of these two types of models for well-represented chemical spaces were below 5%. But more recent deep learning architectures such as graph neural networks and transformer models trained on vast mutagenicity datasets have performed better with out-of-domain chemical structures, a vital ability for the structurally varied NDSRIs that are becoming a flood during the current regulatory emergency [65]. The MELLODDY federated learning project showed that the privacy-preserving collaborative training of 10 pharmaceutical companies led to QSAR models with predictive performance equal to or better than those developed locally within companies, highlighting the possibility of implementing industry-wide data sharing forms in impurity mutagenicity prediction. As tools for visualization of models, like SHAP-based substructure attribution, become a standard regulatory submission, predictive models based on AI are likely to be increasingly accepted. Data independent acquisition (DIA) in high-resolution mass spectrometry is a very promising analytical development. Data Independent Acquisition (DIA) is different from data dependent acquisition in that it fragments all precursor ions, not just the most abundant ones, thereby allowing the identification of trace level impurities that were not initially known at the time of analysis. This is a game-changer for stability indicating method development, as degradation products at very low levels that can only be detected later when samples are reanalysed against expanded spectral libraries during long-term storage. The incorporation of green analytical chemistry principles, which are now measurable (e.g., AGREE score, National Environmental Method Index pictogram, and Green Analytical Procedure Index) into impurity method development is a regulatory expectation that is becoming increasingly codified. The reduction of solvents, elimination of toxic mobile phase additives and minimization of sample preparation steps are all environmental and regulatory compliance issues that are critical to designing impurity profiling methods without compromising sensitivity [66].
CONCLUSION
The body of evidence reviewed in this paper demonstrates that the practice of impurity control in oral solid dosage forms has moved past the analytical problem-solving approach and is now seen as a science that must be learned in combination with the disciplines of solid state physical chemistry, excipient reactivity science, toxicologic risk assessment, and regulatory lifecycle management. The ICH Q3 threshold system is an indispensable and scientifically sound basis, but the nitrosamine crisis has clearly shown that there are structurally significant areas of blind spots, namely the lack of prediction of drug product-level reactivity between the nitrite in the excipients and susceptible API amine groups, that were revealed not by inadequate analytical methods but by inadequate chemical risk imagination during the formulation process. What is most important for the field is not just the faster and more sensitive detection, important as those factors are, but the systematic inclusion of predictive chemistry in the formulation design process, so that excipient nitrite burden, peroxide content, reducing sugar content, pH microenvironment are considered as impurity risk factors along with the traditional compatibility end points. With federated machine learning models for mutagenicity prediction coming to maturity, and data-independent acquisition HRMS now allowing for retroactive detection of degradation events that were previously undetected using targeted analyses, the field is gaining the tools to implement true prospective impurity governance and the greatest need of the pharmaceutical sciences community is to integrate the tools into regulatory submissions, before the impurities reach the patient.
REFERENCES


Samruddhi Gaikwad, Kiran Shinde, Impurity Profiling and Control Strategies in Oral Solid Dosage Forms: Advances in Detection, Regulatory Thresholds and ICH-Guided Quality Assurance, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 3732-3751. https://doi.org/10.5281/zenodo.21436600
 10.5281/zenodo.21436600
10.5281/zenodo.21436600