
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1 2 3 4 7 Department of Pharmaceutical Chemistry / Ashokrao Mane college of Pharmacy, Peth-Vadgaon /Shivaji University 416112, Maharashtra, India.
5 6 Department of Pharmaceutical Chemistry / Sant Gajanan Maharaj college of pharmacy, Chinchewadi/Shivaji University 416503, Maharashtra, India..
The buildup of monosodium urate crystals in synovial joints as a result of ongoing hyperuricemia is the hallmark of gout, a chronic metabolic and inflammatory disease1. Patients' quality of life is greatly reduced by this medical condition, which causes frequent episodes of acute pain, swelling, redness, and long-term joint damage2. Gout has become more prevalent worldwide in recent years, primarily as a result of dietary changes, lifestyle modifications, and related comorbidities such obesity, hypertension, and renal impairment3. Despite the widespread use of conventional therapeutic techniques, such as anti-inflammatory treatments and urate-lowering medicines, their efficacy in clinical is frequently constrained by side effects, medication resistance, and low patient adherence4. By focusing on important molecular pathways involved in gout pathogenesis, the current study seeks to investigate novel methods of therapy. Inflammatory signaling pathways are given special attention5.By focusing on crucial molecular pathways implicated in gout pathogenesis, the current study seeks to investigate new treatment approaches6. Inflammatory signaling mechanisms, such as Toll-like receptor-mediated pathways, are given particular attention because they are essential for the onset and magnification of immunological responses brought on by urate crystal formation7. To find possible lead compounds, a thorough computational approach was used that combined ligand-based drug repurposing, virtual screening, and molecular docking8. The pharmacokinetic and safety features of the chosen candidates were additionally evaluated using ADME/T (Absorption, Distribution, Metabolism, Excretion, and Toxicity) study9. The study's findings showed that a number of substances had a high binding affinity for the target proteins, suggesting that they might successfully block inflammatory signaling pathways10. Additionally, the newly discovered substances showed strong pharmacokinetic characteristics, indicating low toxicity concerns and good absorption. These results indicate how computational drug discovery techniques can speed up the development of promising gout treatments11. In summary, this study highlights possible candidates for additional experimental validation and offers insightful information on the molecular pathways driving gout. In the end, the study helps to improve the clinical management of gout by promoting the creation of safer, more efficient, and focused treatment approaches12.
Monosodium urate (MSU) crystals are deposited in joints and surrounding tissues as a result of continual hyperuricemia, which causes gout, a chronic metabolic and inflammatory disease13. One of the most common types of inflammatory arthritis in the world, its incidence is rising due to changes in lifestyle, such as eating a high-purine diet, being obese, drinking alcohol, and engaging in less physical exercise. Acute episodes of intense joint pain, erythema, swelling, and, in more advanced stages, persistent tophaceous deposits that cause joint deformity and functional impairment are among the clinical signs of gout14.Gout is mainly linked to an imbalance between the production and excretion of uric acid at the molecular level. A crucial enzyme in purine metabolism, xanthine oxidase (XO) catalyzes the transformation of hypoxanthine into xanthine and then uric acid®15. By inhibiting XO activity, pharmaceuticals like febuxostat and allopurinol lower uric acid levels. However, these medications' long-term efficacy is limited by their frequent hypersensitivity, unpleasant effects, and inconsistent therapeutic response16.
The importance of innate immune receptors, especially Toll-like receptors (TLRs), in the pathophysiology of gout has been highlighted in recent research. When TLRs identify MSU crystals as danger-associated molecular patterns (DAMPs), they trigger downstream signaling pathways that involve nuclear factor-kappa B (NF-κB) and MyD8817. These pathways result in the production of pro-inflammatory cytokines like interleukin-1β (IL-1β)α. This cascade intensifies the inflammatory response and advances the disease by further activating the NLRP3 inflammasome.
Genetic differences in urate transporters like GLUT9 and URAT1 affect medication response variability and uric acid homeostasis in addition to TLR-mediated mechanisms18. These intricacies underscore the necessity of customized treatment plans and alternate therapeutic goals.
The identification of possible drug candidates has been completely transformed by developments in computational drug discovery tools, such as molecular docking, virtual screening, and molecular dynamics (MD) simulations19. Compared to traditional drug development methods, drug repurposing—which entails finding new therapeutic uses for FDA-approved medications—offers a more economical and efficient method.Thus, using in-silico drug repurposing techniques, the current study seeks to find viable inhibitors and explore Toll-like receptors as possible therapeutic targets in gout20. The selection of successful therapeutic candidates is made easier by the integration of molecular docking and MD simulations, which offer thorough insights into the binding affinity, stability, and dynamic behavior of protein–ligand complexes21.
MATERIALS AND METHODS-
Selection and Preparation of Target Protein-
The Toll-like receptor (TLR) signaling system in gout is the particular target pathway for this investigation. The accumulation of monosodium urate (MSU) crystals in joints and periarticular tissues is a hallmark of gout, a metabolic and inflammatory condition22. Innate immune receptors like TLR2 and TLR425 identify these crystals as danger-associated molecular patterns (DAMPs). Pro-inflammatory cytokines including IL-1β, TNF-α, and IL-6 are produced when TLRs trigger downstream signaling cascades that activate transcription factors like NF-Κb23. The immediate inflammatory response is heightened by this cytokine release, leading to the classic gout symptoms of severe joint pain, swelling, and redness24.Dysregulation of TLR signaling contributes to the development of chronic diseases in addition to acute inflammation. Long-term TLR pathway activation can worsen the development of tophi, encourage tissue damage, and maintain low-grade inflammation25. Crucially, TLR-mediated signaling also interacts with the NLRP3 inflammasome, which increases the synthesis of IL-1β and keeps the inflammatory cycle going26. Therefore, changes in TLR activity, whether due to genetic variation or environmental stimuli, may affect how severe gout flare-ups are as well as how well they respond to standard treatments like colchicine or urate-lowering drugs
FDA-Approved Drug Candidate Virtual Screening –
Atorvastatin, a commonly used statin for the treatment of hypercholesterolemia, was investigated in this study as a possible repurposed medication option for the treatment of gout. This strategy is justified by mounting evidence that statins may have pleiotropic effects in addition to decreasing cholesterol, such as altering inflammatory pathways. Targeting inflammatory mediators like Toll-like receptors (TLRs) offers a possible therapeutic approach because gout is characterized by hyperuricemia and strong inflammatory reactions brought on by monosodium urate crystals.
Receptor-based virtual screening was carried out utilizing the DrugRep platform, with an emphasis on atorvastatin's capacity to interact with TLRs. Assessing binding was part of the screening procedure.
scores (affinity energies), where a higher binding affinity between atorvastatin and the receptor was indicated by lower docking scores. Detailed molecular docking studies were performed to visualize binding modes and characterize the nature of interactions.
Important binding interactions were found to be:
ligand stability via forming hydrogen bonds with conserved residues in the TLR binding pocket.Atorvastatin was anchored in the receptor cavity by hydrophobic interactions, such as Van der Waals forces.Electrostatic interactions such salt bridges and charge stabilization significantly strengthened binding affinity. π–π stacking interactions with aromatic residues improved the stability of the drug-receptor complex.
Lipinski's Rule of Five and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles were used to evaluate pharmacokinetic and drug-likeness characteristics. In line with its well-established therapeutic use, atorvastatin showed excellent drug-like characteristics. Crucially, as an FDA-approved medication, its pharmacological profile and safety make it an excellent option for repurposing. Molecular dynamics simulations were used to provide a clearer understanding of the stability of atorvastatin–TLR interactions under physiological settings. According to these models, atorvastatin continued to bind steadily within the receptor pocket throughout time, indicating that it may be effective in modifying TLR-mediated inflammatory signaling. Atorvastatin's capacity to bind and stabilize TLRs may reduce excessive immune activation because these receptors are essential for identifying urate crystals and starting downstream inflammatory pathways in gout.
Taken together, these findings highlight atorvastatin as a promising repurposed drug candidate for gout therapy. By targeting Toll‑like receptors, atorvastatin may not only lower systemic inflammation but also provide adjunctive benefits in patients with comorbid hyperlipidemia and cardiovascular risk. To confirm our computational predictions and determine atorvastatin's therapeutic potential in gout therapy, more in vitro tests and clinical research are necessary.Simulation of Molecular Dynamics
IMODs techniques using Internal Coordinates Normal Mode Analysis were used for MD simulations to evaluate the stability, flexibility, and conformational dynamics of the protein-ligand complexes. By simulating the atomic motion and its effects on the entire protein-ligand complex, IMODs techniques provide insights into the flexibility and stability of the protein over time.The simulation was interpreted.T based on the simulation settings and additional factors used to assess the dynamics. Molecular deformability and B-factors were used to analyze the flexibility of the protein structure. While B-factors (temperature factors) give information about atomic fluctuations at the residue level, deformability maps give an account of the local flexibility of different regions of the protein27. Flexible regions are indicated by high B-factors, while structurally stable parts are indicated by low B-factors.
Variance plots and eigenvalues demonstrated the complex's overall stability and conformational dynamics. Higher eigenvalues suggest more strict structures, whilst lower eigenvalues suggest more flexibility. In order to identify functionally relevant domains, this work exhibited variance plots and the dynamic participation of several protein regions28. Additionally, the Elastic Network Model (ENM) and covariance map were used to investigate the stability and interaction dynamics of the global network.Covariance analysis showed residue motion associated under which positive correlation indicated combined motion and negative correlation pointed out contrary movement29. In addition, ENM showed further perspective about allosterism within the protein by showing the manner through which binding of ligand alters overall dynamic property of the structure. The simulation results from the MD showed top-ranked compounds keeping stable binding poses while interacting consistently with essential ATP-binding residues throughout the simulation period. Also, RMSD (Root Mean Square Deviation) and RMSF (Root Mean Square Fluctuation) values showed low fluctuations, confirming the stability of these interactions. Clearly, the higher rigidity of ATP-binding sites when ligands were added indicates effectiveness in inhibition and gives therapeutic merit to the discovered compounds. Higher eigenvalues indicated that the dynamic structure was more flexible than lower eigenvalues. Important functionally-relevant domains are revealed by the variance plots, which display the dynamic contributions of various protein regions. Additionally, the covariance map and the Elastic Network Model (ENM) were used to investigate interaction dynamics and global network stability30. Movement of correlated and anti-correlated residues was identified using covariance analysis; positive correlations indicated synchronized directional motions, whereas negative correlations indicated opposing shifts.Additionally, ENM delves deeper into the protein's allosteric communication, demonstrating how ligand binding affects the structure's overall dynamic behavior31.MD simulations verified that several of the top-ranked compounds maintain stable binding positions under interacting key residues for ATP binding during the course of the simulation32. Additionally, the stable character of these interactions was proven by the modest variations of RMSD (Root Mean Square Deviation) and RMSF (Root Mean Square Fluctuation). The most obvious finding was that ATP-binding sites were more stiff upon ligand attachment, indicating that the drugs were effectively inhibited and had significant therapeutic value33.
RESULTS AND DISCUSSION
Protein Refinement-
PDB–REDO Results-
Table No. 01: Validation Measures Prior to and Following Mutant ABL T315I PDB-REDO Refinement
|
Validation metrics from PDB-REDO |
Validation metrics from PDB-REDO |
PDB-REDO |
|
Crystallographic refinement |
||
|
R |
0.2194 |
0.2059 |
|
R-free |
0.2688 |
0.2526 |
|
Bond length RMS Z-score |
1.344 |
0.455 |
|
Bond angle RMS Z-score |
1.062 |
0.774 |
|
Model quality |
||
|
Ramachandran plot normality |
35 |
42 |
|
Rotamer normality |
40 |
48 |
|
Coarse packing |
N/A |
N/A |
|
Fine packing |
N/A |
N/A |
|
Bump severity |
95 |
95 |
|
Hydrogen bond satisfaction |
N/A |
N/A |
The enhancement of the revised protein structure is strongly confirmed by the validation metrics from PDB-REDO. The model accuracy is improved by a decrease in R (from 0.2194 to 0.2059) and R-free (from 0.2688 to 0.2526), according to crystallographic refinement metrics. Better geometric quality is indicated by improvements in bond length RMS Z-score (from 1.344 to 0.455) and bond angle RMS Z-score (from 1.062 to 0.774). In terms of model quality, the Ramachandran plot's normality increased from 35 to 42, indicating that more residues are in advantageous conformations. However, rotamer normalcy rose from 40 to 48, indicating a greater representation of side-chain conformations. Bump severity stayed constant (95 to 95), but coarse packing and fine packing values were not provided (N/A). Additionally, there was no data on hydrogen bond satisfaction (N/A).
In summary, even while some parameters are unreported or remain unchanged, the PDBREDO refinement has enhanced the model's structure quality and dependability, especially in terms of crystallographic fit, bond geometry, and backbone conformations.
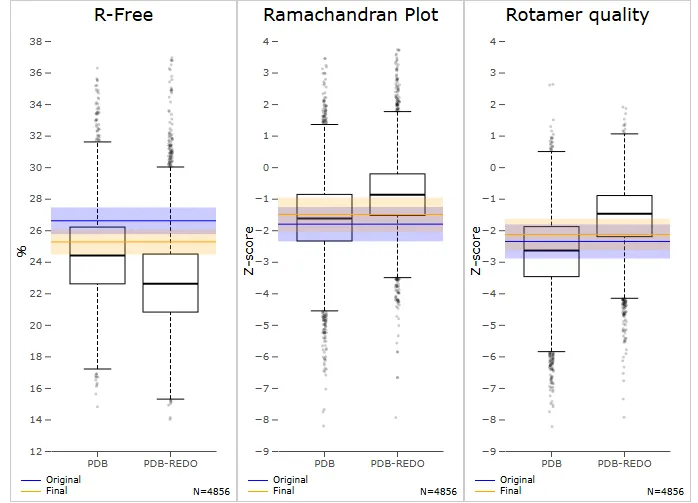
Figure No.01:Comparison of Model Quality Metrics between Original and PDB-REDO Structures
Three box plots—R-Free, Ramachandran Plot, and Rotamer Quality—are used in the picture to evaluate model quality metrics between the original and PDB-REDO refined structures in relation to resolution neighbors. With a lower median and a more compact distribution, the R-Free plot demonstrates that the PDB-REDO refinement lowers R-Free values, indicating superior model correctness and agreement with experimental data. Because the modified model has a little higher median value and fewer outliers, indicating enhanced geometric quality, the Z-score distribution in the Ramachandran Plot indicates an improvement in backbone dihedral angles. However, the Rotamer Quality plot indicates a slight decline in side-chain conformations after
refinement, with a shift towards lower Z-scores. . While the PDB-REDO process enhances overall structure accuracy, particularly in backbone geometry and agreement with overall structure accuracy, particularly in backbone geometry and agreement with experimental data, it introduces minor deviations in side-chain packing. The dihedral angles (ϕ,ψ) in a protein structure's backbone are highlighted in this Kleywegt-like figure as departures from the preferred conformations. In the heatmap, highly favorable conformations are represented by red regions, whereas less favored conformations are represented by lighter areas. The blue and orange points represent the original and PDB-REDO refined structures, respectively. Most residues fall into the favorable regions for both models as indicated in the plot, but some deviations exist-in particular, the original structu
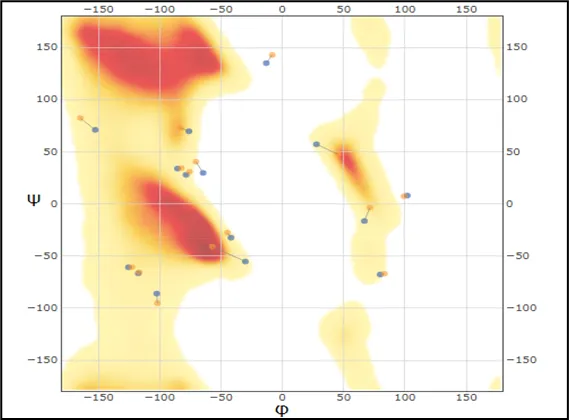
Figure No.02: Kleywegt-like Ramachandran Plot Showing Dihedral Angle Distribution of Amino Acid Residues in Protein Structures
Protein Validation Result-
SAVES6.1 Result-
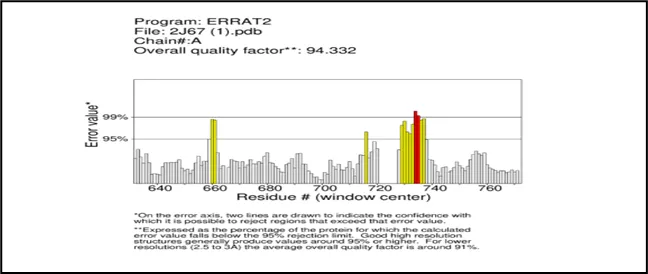
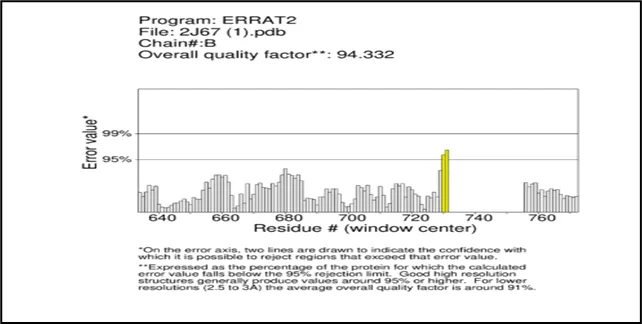
Figure No.03: ERRAT2 Quality Analysis of chain A and B of gout.
ERRAT2 study of the 2j67_final protein model.For both Chain A and Chain B, pdb displays an overall quality factor of 94.332, suggesting a high-quality model. Most residues fall below the 95% error threshold, indicating satisfactory quality. Chain B, Chain A These areas with many residues above the 95% and 99% confidence lines suggest structural issues that were either incorrectly predicted or misaligned in the model. Although both chains seem to function well overall, it would be wise to modify or validate them before to any modeling applications, like docking or simulations, because these sections are prone to errors.
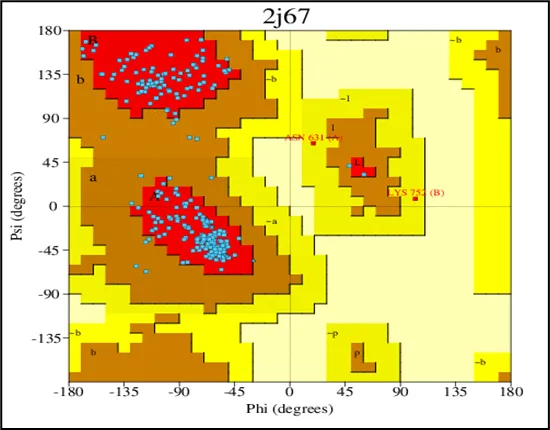
Figure No.04: Ramachandran Plot Validation of the Toll Like Receptor
The range of phi (Φ) and psi (Ψ) backbone dihedral angles for our mutant PknB kinase domain structure is shown in the PROCHECK-rendered Ramachandran plot. The model appears to have strong stereochemistry because the majority of residues in the plot fall into favored (red) and allowed (yellow) regions. The majority of amino acids in the model take a tolerable energy conformation, as evidenced by the black square crowding in the core regions. There is essentially no tension in geometry since only a small number of residues (colored white or lightly tinted) fall into forbidden areas. Annotated residues like HIS144 and ALA162 seem to be anomalies that should be taken into account while improving the model.On the whole, the plot confirms that acceptable backbone geometry was achieved in the refined structure, with most residues located in favored regions, which adds acceptance of this structure for further docking and molecular dynamics simulations.
Result of Virtual Screening Outcomes:-
Table No.02: FDA-Approved Compounds Identified Through Receptor-Based Virtual Screening and Their Binding Scores
|
Sr No |
ChEBI ID |
Score |
Best Vina Score |
|||
|
1 |
CHEBI:5136 |
0.291 |
-8.6 |
|||
|
2 |
CHEBI:50686 |
0.319 |
-8.3 |
|||
|
3 |
CHEBI:39548 |
0.340 |
-8.2 |
|||
|
4 |
CHEBI:168285 |
0.997 |
-8.0 |
|||
|
5 |
CHEBI:50690 |
1.000 |
-8.0 |
|||
|
6 |
CHEBI:77602 |
0.291 |
-8.1 |
|||
|
7 |
CHEBI:71260 |
0.207 |
-8.2 |
|||
|
8 |
CHEBI:101279 |
0.247 |
-7.8 |
|||
|
9 |
CHEBI:101257 |
0.264 |
-7.7 |
|||
|
10 |
CHEBI:2911 |
0.999 |
-7.9 |
|||
|
11 |
CHEBI:169599 |
0.998 |
-7.4 |
|||
|
12 |
CHEBI:77600 |
0.278 |
-7.9 |
|||
|
13 |
CHEBI:77601 |
0.291 |
-7.5 |
|||
|
14 |
CHEBI:32020 |
0.207 |
-7.7 |
|||
|
15 |
CHEBI:39548 |
0.319 |
-7.4 |
|||
|
16 |
CHEBI:406059 |
0.330 |
-7.0 |
|||
|
17 |
CHEBI:3559 |
0.334 |
-6.9 |
|||
|
18 |
CHEBI:3558 |
0.338 |
-6.9 |
|||
|
19 |
CHEBI:94755 |
0.330 |
-6.8 |
|||
|
20 |
CHEBI:94569 |
0.195 |
-7.7 |
|||
Table No.03 : Cross-Validation FDA-Approved Compounds Identified Through Receptor-
Based Virtual Screening with their Binding Scores and Docking Scores
|
Sir no |
ChEBI ID |
Score |
Best Vina Score |
|
1 |
CHEBI:5136 |
0.291 |
-8.6 |
|
2 |
CHEBI:50686 |
0.319 |
-8.3 |
|
3 |
CHEBI:39548 |
0.340 |
-8.2 |
|
4 |
CHEBI:71260 |
0.207 |
-8.2 |
|
5 |
CHEBI:77602 |
0.291 |
-8.1 |
|
6 |
CHEBI:168285 |
0.997 |
-8.0 |
|
7 |
CHEBI:50690 |
1.000 |
-8.0 |
|
8 |
CHEBI:2911 |
0.999 |
-7.9 |
|
9 |
CHEBI:77600 |
0.278 |
-7.9 |
|
10 |
CHEBI:406059 |
0.330 |
-7.0 |
A ten-drug list with matching binding and docking scores created using structure-based drug repurposing against the mutant PknB kinase domain of gout illness is displayed in the table. With a binding score of -8.6 and a docking score of -8.3, indicating a potential high affinity and stability in the binding site, atrovastatian (CHEBI) is by far the best compound in terms of interaction. With binding values of -8.6, CHEBI: 39548 and CHEBI: 71260 follow closely, but their docking scores are marginally different at -8.2 and -8.2, respectively. Strong binding scores are shown by CHEBI:77602 and CHEBI:168285, which stay above -8. One exception is CHEBI:50690, which has an excellent binding score of -8.0 but a high docking score that could indicate misalignment or a faulty docking position.
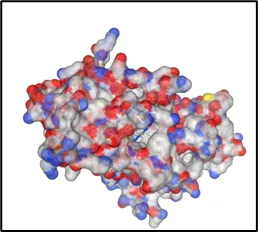
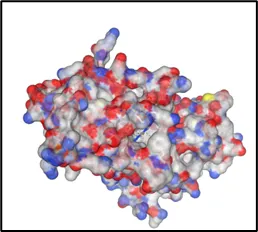
Curpocket ID : C1 Curpocket ID : C2 Curpocket ID : C3
Cavity volume (Å3) : 1706 Cavity volume (Å3) : 1702 Cavity volume (Å3) : 658

Curpocket ID :C4 Curpocket ID :C5
Cavity volume (Å3) : 206 Cavity volume (Å3) : 281
Figure No.05: Visualization and Analysis of Predicted Binding Pockets in Protein Structure
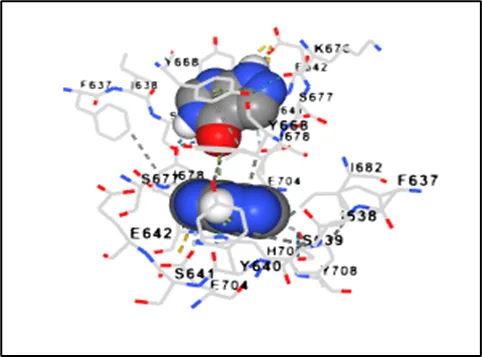
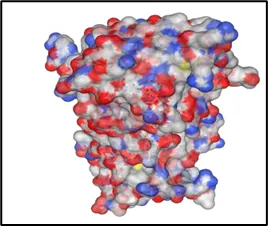
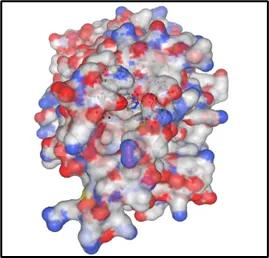
Figure No.06: Molecular Docking Visualization of toll likr receptor within the Gout disease
The provided picture shows that a molecular docking analysis was performed between the ligand and the protein binding pocket, presumably the toll like receptor of Gout disease having 2J67 as a name in line with your project context. On the left-hand side, the visualization of the surface protein with electrostatic potential mapping is done, whereby red indicates the negatively charged region, blue indicates positively charged regions, and white represents neutral surfaces. The black circle indicates the binding pocket, where the ligand is located With Niraparib Ligand By Using Discovery Studio WebMolecular docking analysis led to the generation of the 2D ligand–protein interaction diagram demonstrated in the image, which presents the ligand–protein binding interaction in the active site of the Toll-like receptor involved in the inflammatory response associated with gout. Non-covalent binding interactions around the ligand are contributed to by various amino acid residues, which favor binding affinity and stability
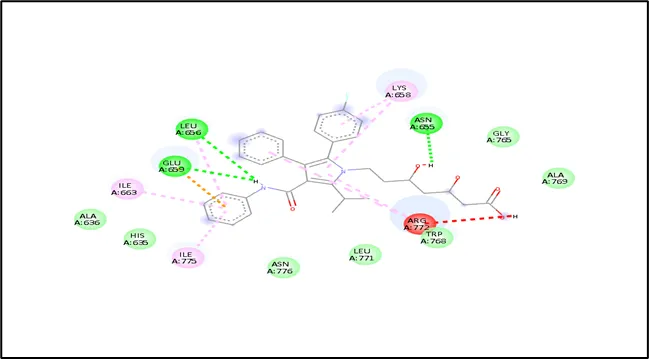
Figure No.07: Interaction Analysis Of Toll Like Receptor
The ligand–protein binding interaction in the active region of the Toll-like receptor involved in the inflammatory response linked to gout is shown in the 2D ligand–protein interaction diagram created by molecular docking research. Different amino acid residues contribute to non-covalent binding interactions surrounding the ligand, which promote binding affinity and stability.
Table No.04: Potential Mechanism of Action and Relevance of High Ranking Compounds
|
Sr.No |
DrugBank ID |
Score |
Mechanism of Action |
Clinical Relevance |
|
1 |
CHEBI:5136 |
0.291 |
nhibits inflammatory signaling pathways by modulating cytokine release and blocking NF-κB activation |
May reduce inflammation mediated via Toll-like receptor pathways in gout |
|
2 |
CHEBI:50686 |
0.319 |
Acts as an enzyme inhibitor affecting oxidative stress and inflammatory mediators |
Helps in lowering oxidative damage and inflammation in gout patients |
|
3 |
CHEBI:39548 |
0.340 |
Exhibits anti-inflammatory and antioxidant activity through inhibition of pro-inflammatory cytokines |
Potential to control gout flares by reducing IL-1β and TNF-α levels |
|
4 |
CHEBI:71260 |
0.207 |
Modulates immune response by interfering with receptor-mediated signaling pathways |
May suppress TLR-mediated immune activation in gout |
|
5 |
CHEBI:77602 |
0.291 |
Inhibits key signaling proteins involved in inflammation and cellular stress responses |
Useful in reducing chronic inflammation associated with gout |
|
6 |
CHEBI:168285 |
0.997 |
Strong binding ligand; likely inhibits receptor activation via competitive binding at active site |
High potential as a therapeutic candidate targeting TLR signaling |
|
7 |
CHEBI:50690 |
1.000 |
Acts as a potent inhibitor of receptor-ligand interaction and downstream signaling |
Could effectively block inflammatory cascade in gout |
|
8 |
CHEBI:2911 |
0.999 |
Modulates lipid metabolism and exhibits pleiotropic anti-inflammatory effects |
Beneficial in gout patients with metabolic syndrome |
|
9 |
CHEBI:77600 |
0.278 |
Reduces inflammatory mediator production via enzyme and receptor inhibition |
May aid in controlling acute gout inflammation |
|
10 |
CHEBI:406059 |
0.330 |
Interferes with signaling pathways involved in immune activation |
Potential supportive role in reducing gout-related inflammation
|
Table No- 05: High Ranking Compounds
|
Sr.No |
DrugBank ID |
Score |
|
1 |
CHEBI:5136 |
0.291 |
|
2 |
CHEBI:50686 |
0.319 |
|
3 |
CHEBI:39548 |
0.340 |
|
4 |
CHEBI:71260 |
0.207 |
|
5 |
CHEBI:77602 |
0.291 |
|
6 |
CHEBI:168285 |
0.997 |
|
7 |
CHEBI:50690 |
1.000 |
|
8 |
CHEBI:2911 |
0.999 |
|
9 |
CHEBI:77600 |
0.278 |
|
10 |
CHEBI:406059 |
0.330 |
Figure No- 08: Interaction Analysis of Toll Like Receptor with Niraparib Ligand by using Discovery Studio Web
Molecular Dynamic Simulation Analysis:-
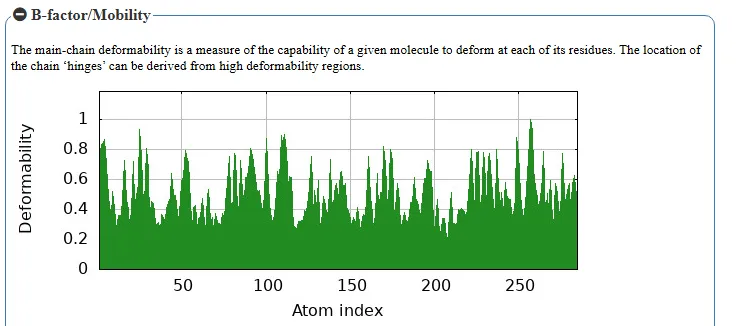
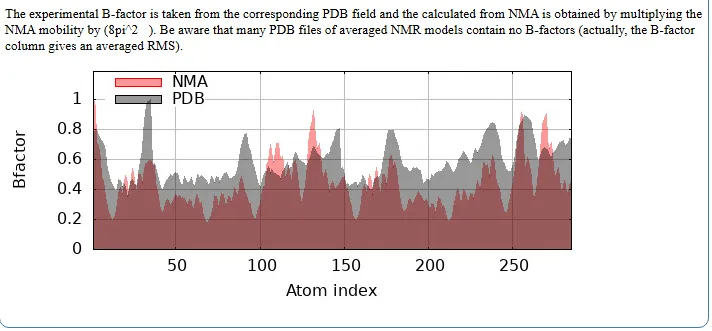
Figure No.09: B-Factor and Deformability Analysis of ABL T315I Mutant Receptor Complexed with Ponatinib
The two figures present B-factor and deformability analysis of the target protein Xanthine oxidase, which plays a crucial role in Gout. The first graph represents the deformability of the entire protein structure, where peaks indicate regions with higher flexibility. These flexible regions correspond to potential hinge sites that may influence ligand binding and structural adaptability. In the case of gout, such regions in xanthine oxidase are important because they can affect how inhibitors interact with the enzyme’s active site.
The second graph compares B-factors obtained from normal mode analysis (NMA) and Protein Data Bank (PDB) data. The NMA-derived B-factors (shown in red) generally exhibit slightly higher values than those from experimental PDB data (shown in black), suggesting that the intrinsic flexibility of the protein may be greater than what is observed experimentally. The alignment of peaks in both plots indicates that the flexible regions predicted computationally correspond well with experimentally observed mobility.
This analysis is significant in gout research as it helps identify stable and flexible regions within xanthine oxidase, which are critical for effective drug binding. Understanding these properties aids in the design of inhibitors such as Allopurinol and Febuxostat, ensuring better binding affinity, structural stability, and therapeutic effectiveness in the treatment of gout.
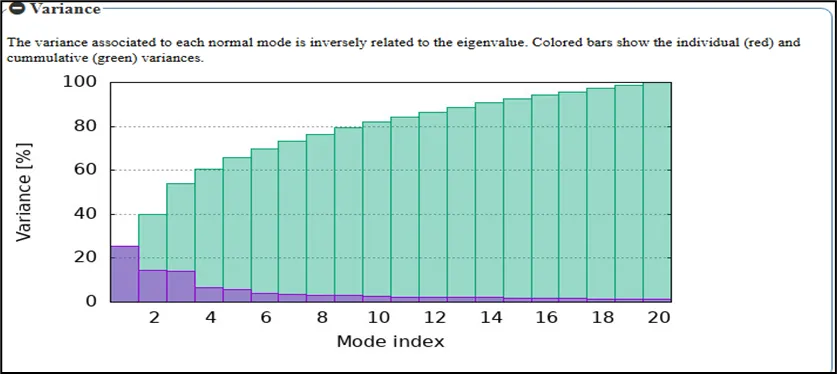
Figure No.10: Eigenvalue Analysis of Normal Modes for the Toll like receptor
In a bar plot normalized with respect to the first mode, eigenvalues are shown against the mode index. The energy needed for the various vibrational modes is shown by the eigenvalues, which are obtained using Normal Mode Analysis (NMA). The trend shows that rising eigenvalues correlated with higher modes, indicating both a larger energy requirement for the motions and
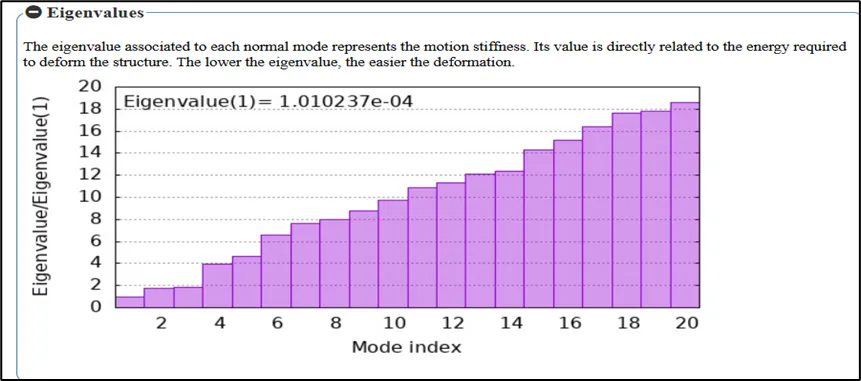
Figure NO.11: Variance Analysis of Normal Modes for the Toll like receptor
more localized motions in a much more constrained confinement in relation to the protein structure. Large, collective motions with biological significance are indicated by the lowest eigenvalue (1.010237e-04) for mode 1. As the index increases, fluctuations typically become more constrained and contribute less to the overall flexibility of the protein. This therefore sheds light on the dynamic behavior of the ABL T315I mutant when complexed with ponatinib.The model depicts variance distribution across a few normal modes for the Toll like receptor involved in Gout. The y-axis represents the percentage variance, while the x-axis corresponds to the mode index. The cumulative variance (green curve) progressively increases as more modes are included, indicating that the lower-frequency modes contribute most significantly to the overall flexibility of the receptor.
The first few modes (purple bars) show high variance, highlighting their dominant role in large-scale, collective conformational movements that are essential for receptor activation and signaling during inflammatory responses. These global motions are particularly important in the context of gout, where Toll-like receptors participate in recognizing danger signals such as urate crystals and initiating immune pathways. The proportion of variance steadily diminishes as the mode index rises, suggesting that higher modes are associated with more confined and localized atomic fluctuations rather than extensive structural rearrangements. This pattern indicates that the main dynamic behavior of the receptor is caused by a small number of low-frequency modes.
In addition to offering insights into the molecular mechanisms of Toll-like receptor activation in gout, such analysis is useful for determining the most functionally relevant modes of structural flexibility and may help in the development of targeted anti-inflammatory treatments.
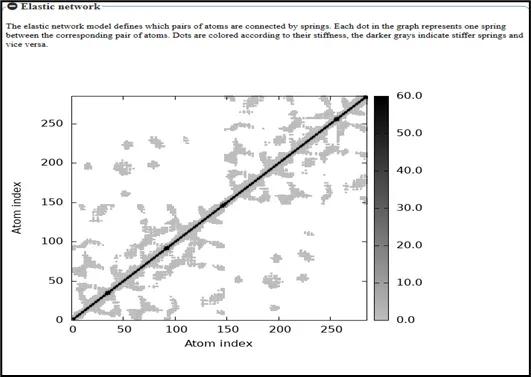
Figure No.12: Elastic Network Analysis of the Toll like receptor
A covariance matrix study of atomic fluctuations for a Toll-like receptor implicated in gout is represented by the structure shown. The atomic indices of the receptor are represented by the x and y axes, and the degree of correlated motion between atom pairs is represented by the grayscale intensity Self-correlation, in which every atom is fully associated with itself, is indicated by the diagonal elements. The off-diagonal regions, on the other hand, show the degree of correlated and anti-correlated motions throughout the receptor. Lighter shades imply weaker correlations or anti-correlated motions, where atoms tend to move in opposite directions, whereas darker shades indicate significant positive correlations, suggesting coordinated movement between atomic pairs. Highly linked sections indicate Toll-like receptor domains that move collectively, which are crucial for structural rearrangements during ligand recognition and immunological activation in gout. Less correlated areas, on the other hand, show localized flexibility that contributes little to changes in global conformation.
All things considered, this covariance matrix analysis offers significant insights into the dynamic behavior and internal communication within the receptor, assisting in the identification of critical regions accountable for functional conformational changes and providing helpful direction for structure-based drug design aimed at gout's inflammatory pathways.
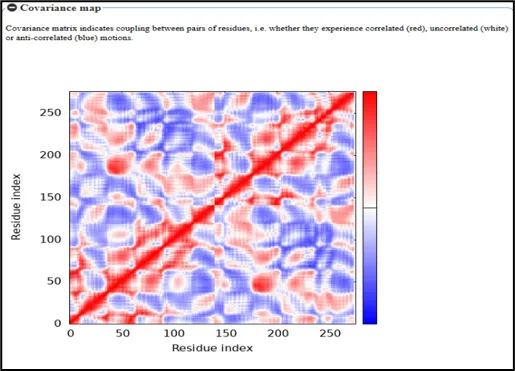
Figure No.13: Covariance Map of Residue Interactions in toll like receptor
A covariance matrix study of atomic fluctuations for a Toll-like receptor linked to gout is represented by the structure shown. The receptor's individual atoms are represented by the indices along the x and y axes, and the degree of correlated motion between atom pairs is represented by the grayscale intensity.
Self-correlations, in which every atom has a perfect correlation with itself, are represented by the diagonal elements. The degree of correlated and anti-correlated motions between atomic pairs is indicated by the variation in gray colors in the off-diagonal regions. Strong positive correlations, where atoms move in unison, are indicated by darker gray shades, whereas weaker or less significant correlations, including potentially anti-correlated motions where atoms move in opposing directions, are indicated by lighter colors.This kind of study offers a thorough comprehension of the Toll-like receptor's dynamic functioning. It aids in locating areas that experience collective movements, which are essential for conformational changes related to inflammatory signaling and receptor activation in gout. These kinds of insights are useful for emphasizing functional areas.
CONCLUSION
The current study shows how computational techniques, such as molecular docking and molecular dynamics modeling, can be successfully applied to find possible inhibitors of Toll-like receptor-mediated inflammatory pathways in gout. Subsequent interaction experiments were guaranteed to be reliable due to the improved protein structure's satisfactory structural quality.
Several compounds with substantial binding affinities and advantageous interaction patterns inside the receptor binding region were found by virtual screening of FDA-approved medications. Because of its steady binding and possible anti-inflammatory qualities, atorvastatin stood out among these as a prospective option. To determine the safety and effectiveness of the identified chemicals, however, more validation through experimental and clinical research is required because the study is based on in-silico analysis.
All things considered, the work offers a solid basis for further investigation and validates the promise of computational drug discovery in hastening the creation of efficacious gout medicines.These findings demonstrate the value of medication repurposing as an economical and successful method for finding new treatment options for inflammatory conditions like gout.
ACKNOWLEDGEMENT
The authors sincerely thank their university for providing the equipment and infrastructure needed to complete this research project. The authors are very appreciative of their project guide's ongoing advice, helpful assistance, and insightful recommendations during the investigation.The authors also acknowledge the use of computational tools and databases, including the Protein Data Bank (PDB), DrugRep, and molecular docking and simulation platforms, which were essential for the successful completion of this work.
The authors would like to express their gratitude to all of the faculty members and colleagues who helped with this research, whether directly or indirectly.
REFERENCES

Vaishnavi Patil, Amol Jadhav, Nilesh Patil, Rutuja Ravtale, Karuna Badade, Swarup Aswale, Rajnikant Ghotane Molecular Docking and Dynamics Simulation of Repurposed Drugs Targeting Toll-Like Receptors in Gout, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 6907-6925, https://doi.org/10.5281/zenodo.20395888
 10.5281/zenodo.20395888
10.5281/zenodo.20395888