
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Rastrasanth College of Pharmacy, Kokamthan, Kopergaon
Lung cancer remains the leading cause of cancer-related mortality worldwide, with therapeutic outcomes often constrained by poor drug selectivity, systemic toxicity, multi drug resistance, and limited intracellular drug accumulation. Nanotechnology-based drug delivery systems have emerged as promising strategies to overcome these limitations and improve therapeutic efficacy. Among various nanocarriers, mesoporous silica nanoparticles (MSNs) have attracted considerable attention owing to their high surface area, tunable pore architecture, excellent biocompatibility, and versatile surface chemistry that enable efficient loading and controlled release of anticancer agents. Recent advances in surface engineering have highlighted the potential of zwitterionic functionalization to impart superior antifouling characteristics, minimize protein corona formation, prolong systemic circulation, and enhance tumor accumulation through improved stealth behavior. Oroxylin A, a naturally occurring flavonoid with significant anticancer activity, has demonstrated promising therapeutic effects against lung cancer; however, its clinical application is limited by poor aqueous solubility and inadequate cellular uptake. The integration of Oroxylin A with zwitterion-functionalized MSNs represents a novel strategy to improve drug bioavailability, intracellular delivery, and cytotoxic efficacy against lung cancer cells. This review comprehensively discusses the pathophysiology of lung cancer, current therapeutic limitations, the principles of nanoparticle-mediated drug delivery, the structural and functional attributes of MSNs, and the biological advantages conferred by zwitterionic surface modification. Furthermore, recent advances in targeted, stimuli-responsive, and theranostic silica-based nanoplatforms are critically evaluated with emphasis on their translational potential for precision oncology. The emerging evidence suggests that zwitterionic MSN systems constitute a promising platform for the development of next-generation nanotherapeutics for effective and targeted lung cancer management.
1.1.Global Burden of Lung Cancer
Lung cancer remains one of the most clinically challenging malignancies of the 21st century, consistently ranking as the leading cause of cancer-related mortality across both developed and developing nations. According to the Global Cancer Statistics compiled by the International Agency for Research on Cancer, lung cancer accounts for approximately 2.2 million new cases annually and is responsible for nearly 1.8 million deaths each year, representing close to 18% of all cancer deaths globally. This alarming epidemiological burden places lung cancer ahead of colorectal, breast, and prostate cancers in terms of mortality, underscoring the urgent need for improved diagnostic and therapeutic strategies. The incidence is particularly high in regions with substantial tobacco consumption, occupational exposure to carcinogens such as asbestos and silica dust, and high levels of air pollution including particulate matter (PM2.5) and indoor biomass smoke—factors of escalating relevance in rapidly industrializing nations including India, China, and Southeast Asian countries.
In the Indian context, lung cancer represents a growing public health crisis. India accounts for a significant proportion of the global lung cancer burden, with incidence rates rising steadily over the past two decades. According to the Indian Council of Medical Research and National Cancer Registry Programme data, lung cancer is among the top five most common cancers in Indian males, and its prevalence among females is also increasing due to indoor air pollution from chulha cooking, passive smoking exposure, and biomass fuel combustion. Urban centers such as Mumbai, Delhi, Chennai, Kolkata, and Pune report significantly higher incidence rates compared to rural regions, largely attributable to vehicular emissions, industrial pollution, occupational exposure, and changing lifestyle patterns. The median age at diagnosis in India tends to be lower than in Western countries, with many patients presenting in their fourth and fifth decades of life, indicating a potentially more aggressive disease course in the genetically and environmentally primed Indian population.
1.2 Clinical Gaps Necessitating Nanomedicine
Late-stage presentation remains a major challenge, with a large majority of Indian patients diagnosed at advanced stages (III/IV) when curative surgical options are limited, resulting in a five-year survival rate of less than 20% in most Indian cohorts. The socioeconomic impact is profound: lung cancer disproportionately affects low- and middle-income populations with limited access to screening programs, specialized oncology care, and expensive targeted therapies. The absence of organized lung cancer screening programs comparable to those in the United States or European Union means that most cases in India are detected incidentally or symptomatically at advanced stages. Efforts to integrate low-dose computed tomography screening into the national health framework are still in early stages, and awareness about risk factors and early warning signs remains inadequate in the general population.
This multifactorial mortality profile defines the rationale for therapeutic innovation through nanomedicine, particularly nanoparticle-based drug delivery systems capable of selective tumor targeting, controlled drug release, and circumvention of biological barriers including protein corona formation, mononuclear phagocyte system clearance, and multidrug efflux.
1.3 The Convergence of Nanomedicine and Natural Product Oncology
Among diverse nanoparticle platforms explored for oncological applications, mesoporous silica nanoparticles occupy a uniquely advantageous position owing to their extraordinarily high surface area (700–1200 m²/g), tunable mesopore diameter, large pore volume, thermal and chemical stability, and well-characterized surface silanol chemistry that facilitates diverse functionalization strategies. Their proven ability to load and retain hydrophobic drug molecules within their mesopore channels makes them particularly suited for compounds such as Oroxylin A, a flavonoid with poor aqueous solubility and low cellular uptake. However, unmodified MSNs suffer from rapid protein corona formation upon entering the bloodstream, which triggers opsonization and accelerated clearance by the mononuclear phagocyte system, severely curtailing tumor accumulation.
To address this limitation, zwitterionic functionalization using sulfobetaine or carboxybetaine silane derivatives is incorporated in the present platform. Zwitterionic coatings generate a tightly bound electrostatically induced hydration layer that resists nonspecific protein adsorption far more effectively than conventional PEG coatings, conferring stealth properties, prolonging systemic circulation, and enhancing passive tumor accumulation through the EPR effect, without triggering anti-coating antibody formation upon repeated dosing he present review integrates these research threads into a coherent analysis of zwitterionic-MSN-mediated Oroxylin A delivery for lung cancer therapy, while contextualizing it within the broader translational nanomedicine landscape.
1.4 Lung Cancer: Epidemiology, Histopathology, and Clinical Challenges
1.4.1 Global and Indian Epidemiology
Lung cancer represents a growing public health crisis globally, with GLOBOCAN statistics highlighting 2.2 million new cases and 1.8 million deaths annually, accounting for close to 18% of all cancer-related deaths worldwide. This positions lung cancer as the single deadliest malignancy, ahead of breast, colorectal, and prostate cancers. In India specifically, the ICMR-NCRP has documented that lung cancer constitutes one of the leading malignancies in males, with rising incidence among females driven by indoor biomass combustion exposure and passive smoking. The Indian demographic burden is compounded by absence of population-wide screening, late presentation, and limited access to advanced therapeutics.
Several well-defined risk factors contribute to lung cancer pathogenesis. Cigarette smoking remains the single largest preventable cause, with tobacco smoke containing over 60 known carcinogens including polycyclic aromatic hydrocarbons (benzo[a]pyrene), nitrosamines, aromatic amines, and benzene derivatives that induce mutagenic DNA damage. Occupational exposures to asbestos, radon, silica dust, diesel exhaust, and certain metals (chromium, nickel, arsenic) increase risk independently and synergistically with smoking. Air pollution, both ambient (PM2.5) and indoor (cooking fumes, second-hand smoke), accounts for an estimated 15–20% of lung cancer cases globally. Genetic predisposition accounts for a smaller but significant fraction of cases, particularly in never-smokers harboring EGFR mutations or ALK rearrangements.
1.4.2 Histological Classification
Lung cancer is classified into two major histological categories based on cell type of origin and biological behavior. Non-small cell lung cancer constitutes approximately 85% of all lung cancer diagnoses and includes three major subtypes.
Adenocarcinoma is the most common subtype, accounting for nearly 40% of all lung cancers. It predominantly arises in peripheral lung regions, is more commonly seen in non-smokers and women, and is strongly associated with EGFR mutations (exon 19 deletions, L858R), ALK rearrangements, ROS1 fusions, and KRAS G12C mutations—making it particularly amenable to targeted molecular therapies.
Squamous cell carcinoma is the second most common subtype, arising centrally near the bronchi. It is more strongly associated with cigarette smoking history and frequently harbors alterations in FGFR1, DDR2, PIK3CA, and PTEN.
Large cell carcinoma represents a diagnosis of exclusion characterized by undifferentiated cells lacking defining features of adenocarcinoma or squamous cell carcinoma, growing and spreading rapidly with poor prognosis.
Small cell lung cancer, accounting for ~15% of lung cancers, is a highly aggressive neuroendocrine tumor almost exclusively associated with heavy cigarette smoking. SCLC is characterized by rapid doubling times (as short as 30 days), early widespread metastasis, and initial chemosensitivity followed by almost inevitable relapse with resistant disease. Unlike NSCLC, SCLC has limited targetable molecular alterations, and treatment options have remained relatively unchanged for decades (platinum-etoposide remains standard). The classification into NSCLC and SCLC has profound implications for treatment selection, clinical trial design, nanoparticle targeting strategies, and prognosis.
1.4.3 Molecular Pathogenesis
The molecular pathogenesis is a complex multi-step process involving sequential accumulation of genetic and epigenetic alterations. KRAS mutations (predominantly at codon 12, G12C, G12D, G12V) are found in 25–30% of NSCLC cases in smokers, resulting in constitutive activation of RAS/MAPK and PI3K/AKT/mTOR signaling cascades driving uncontrolled proliferation. TP53 mutations occur in over 50% of lung cancers, impairing DNA damage response and apoptotic machinery. STK11/LKB1 mutations co-occur with KRAS in a significant subset of cases.
In non-smokers and younger patients, driver mutations more commonly occur in EGFR (10–15% Western NSCLC, 40–50% Asian NSCLC—particularly adenocarcinomas), ALK, ROS1, RET, MET, BRAF, and NTRK. EGFR mutations, particularly exon 19 deletions and L858R point mutations in exon 21, result in ligand-independent receptor activation and downstream proliferative signaling. The discovery of EGFR mutations and subsequent development of EGFR tyrosine kinase inhibitors (gefitinib, erlotinib, afatinib, osimertinib) represented a paradigm shift in advanced NSCLC therapy. Igenetic alterations including DNA methylation (CDKN2A, RASSF1A, DAPK1 promoter hypermethylation), histone modification (H3K27me3, acetylation), and non-coding RNA dysregulation (miR-21, miR-155, let-7 family) play critical roles in lung carcinogenesis. MicroRNA expression profiles are being explored as diagnostic biomarkers (miR-21 in sputum), prognostic indicators, and therapeutic targets (miRNA mimics and antimiRs in clinical development).
1.4.4.Tumor Microenvironment
The tumor microenvironment represents the complex ecosystem surrounding cancer cells, comprising stromal fibroblasts, endothelial cells, immune cells, ECM components, and soluble mediators (cytokines, chemokines, growth factors). The TME actively participates in tumor initiation, progression, immune evasion, and resistance to therapy.
lung cancer, the TME is highly immunosuppressive. Tumor-associated macrophages, predominantly M2-phenotype, secrete immunosuppressive cytokines IL-10 and TGF-β that inhibit cytotoxic T lymphocyte activity and promote tumor angiogenesis and invasion. Myeloid-derived suppressor cells further dampen anti-tumor immune responses via reactive oxygen species production and T cell proliferation suppression. Regulatory T cells accumulate within tumors and suppress effector immune cell function through IL-10 and CTLA-4-mediated mechanisms. The immunosuppressive milieu contributes to the limited efficacy of immunotherapy in approximately 70% of NSCLC patients who derive no long-term benefit from checkpoint inhibition.
Cancer-associated fibroblasts represent another major stromal component that remodels ECM through secretion of collagen, fibronectin, and matrix metalloproteinases (MMP-2, MMP-9), creating a dense physical barrier impeding drug penetration into the tumor core. CAFs also secrete HGF and FGF that activate pro-survival signaling. Elevated interstitial fluid pressure within solid tumors, a consequence of abnormal vasculature and dense ECM, creates a pressure gradient opposing convective drug delivery.
Tumor vasculature in lung cancer is structurally and functionally abnormal: tortuous, leaky blood vessels with irregular blood flow create hypoxic regions and acidosis. Aberrant vasculature with widened interendothelial junctions (200–2000 nm gaps), driven by VEGF overexpression, bradykinin, and nitric oxide, creates size-dependent selective permeability exploited by EPR-mediated nanoparticle accumulation The acidic TME (extracellular pH 6.5–7.0, endolysosomal pH 5.0–5.5) has been exploited for pH-responsive drug release systems, directly relevant to mesoporous silica nanoparticle design.
1.4.5. Treatment Modalities
Surgery remains the cornerstone of curative-intent treatment for early-stage NSCLC (stages I and II). Procedures range from wedge resection and segmentectomy for small peripheral tumors to lobectomy and pneumonectomy for larger or centrally located lesions. Video-assisted thoracoscopic surgery and robotic-assisted thoracic surgery have largely replaced open thoracotomy, offering reduced morbidity, shorter hospital stays, and equivalent oncological outcomes. Only approximately 20–25% of NSCLC patients present with resectable disease at diagnosis, severely limiting surgery's curative applicability.
Platinum-based doublet chemotherapy (cisplatin/carboplatin + paclitaxel, gemcitabine, pemetrexed, or vinorelbine) has long served as backbone treatment for advanced NSCLC. While chemotherapy improves survival and quality of life versus best supportive care, benefits are modest (median OS improvement 2–4 months) and come at the cost of significant systemic toxicity including myelosuppression, nephrotoxicity (cisplatin), neurotoxicity, ototoxicity, and gastrointestinal adverse effects. Resistance, both intrinsic and acquired, remains a major obstacle.
Targeted therapy has been transformed by identification of oncogenic driver mutations. EGFR TKIs (gefitinib, erlotinib, afatinib, osimertinib), ALK inhibitors (crizotinib, alectinib, brigatinib, lorlatinib), and inhibitors targeting ROS1, BRAF V600E, RET, MET exon 14, and KRAS G12C (sotorasib, adagrasib) achieve high response rates and prolonged progression-free survival in biomarker-selected populations. Howevr, resistance inevitably develops through diverse mechanisms including secondary target mutations (T790M in ~50–60% of acquired EGFR TKI resistance, C797S as next-generation mutation), bypass pathway activation (MET amplification, HER2 amplification), histologic transformation (NSCLC to SCLC transformation in ~5–10%), and drug efflux.
Immunotherapy with immune checkpoint inhibitors targeting PD-1/PD-L1 axis—pembrolizumab, nivolumab, atezolizumab, durvalumab—has emerged as a major therapeutic advance. These agents demonstrate durable responses and improved overall survival as monotherapy or combined with chemotherapy. However, only a subset of patients (20–30%) derive meaningful long-term benefit, and predictive biomarkers beyond PD-L1 expression (tumor mutational burden, tumor-infiltrating lymphocytes, gene expression signatures) remain incompletely characterized. Immune-related adverse events (irAEs) can affect virtually any organ system—colitis, hepatitis, pneumonitis, thyroiditis, hypophysitis, myocarditis—ranging from manageable to life-threatening.
1.4.6.Unmet Clinical Needs
Despite advances, advanced lung cancer prognosis remains poor. Severa limitations underscore the urgent need for novel delivery strategies:
These collective limitations highlight the compelling rationale for nanoparticle-based drug delivery systems capable of selective tumor targeting, minimization of systemic toxicity, overcoming drug resistance, and enabling combination with existing therapies for synergistic effects.
2. NANOPARTICLE-BASED DRUG DELIVERY IN CANCER THERAPY
2.1 Fundamentals of Nanomedicine
Nanomedicine represents the application of nanotechnology principles to disease prevention, diagnosis, and treatment, utilizing materials and devices at the nanoscale (1–1000 nm range). Materials at this scale possess unique properties distinct from bulk counterparts: exceptionally high surface area-to-volume ratios, quantum mechanical effects, enhanced reactivity, and tunable optical, magnetic, and electronic characteristics. In cancer therapy, nanomedicine enables precise spatial and temporal control over drug release, improved pharmacokinetic profiles, reduced systemic toxicity, tumor-selective drug accumulation, and theranostic integration.
2.2 Classification of Nanoparticles
Nanoparticle systems for drug delivery are broadly classified by composition into:
Organic nanoparticles include liposomes, polymeric nanoparticles, solid lipid nanoparticles, nanoemulsions, dendrimers, cyclodextrin-based nanoparticles, and protein-based nanoparticles (albumin-bound paclitaxel/Abraxane, FDA-approved 2005). Liposomes carry both hydrophilic drugs in their aqueous interior and hydrophobic drugs in lipid bilayers but suffer physical instability and opsonization-induced rapid clearance.
Inorganic nanoparticles include gold nanoparticles, iron oxide nanoparticles, carbon nanotubes, quantum dots, and silica nanoparticles. Mesoporous silica nanoparticles stand distinct from other inorganic systems through their exceptionally high surface area (700–1200 m²/g), large pore volume, tunable pore size, and versatile silica surface chemistry enabling diverse functionalization strategies.
Hybrid nanoparticles combine organic and inorganic components for synergistic properties, e.g., lipid-coated PLGA-PEG-hybrid systems, lipid-coated silica, polymer-metal organic framework composites.
2.3 Passive and Active Targeting
Two principal targeting strategies underpin nanomedicine:
Passive targeting through the EPR effect exploits the aberrant, leaky vasculature and defective lymphatic drainage of solid tumors, enabling preferential accumulation of nanoparticles in the 10–200 nm size range. The Effect was first described by Maeda and Matsumura in 1986 and has since become the foundational concept for anticancer nanomedicine design. In normal vasculature, tight endothelial junctions restrict macromolecular passage. In tumor vasculature, characterized by widened interendothelial junctions (200–2000 nm gaps), irregular pericyte coverage, and overexpression of vascular permeability factors, preferential accumulation of appropriately sized nanoparticles occurs. Defective lymphatic drainage prevents clearance, prolonging intra-tumor retention.
Active targeting supplements passive accumulation through surface decoration with recognition moieties (monoclonal antibodies, antibody fragments, peptides, aptamers, small molecules) binding receptors overexpressed on cancer cells: folate receptor alpha in ~30–40% of NSCLC, EGFR in 40–80%, transferrin receptor in highly proliferative cells, HER2, integrin αvβ3 in tumor vasculature, CD44 in cancer stem cells. Active targeting primarily enhances intracellular delivery through receptor-mediated endocytosis rather than altering extravasation.
2.4 EPR Effect in Lung Cancer
In lung cancer, EPR-mediated accumulation is complicated by tumor heterogeneity, elevated interstitial fluid pressure, the dense stromal compartment in certain histological subtypes, and unique pulmonary circulation dynamics. Nanoparticle administered intravenously must survive first-pass through pulmonary capillaries before reaching systemic circulation, with significant sequestration possible depending on size, charge, and surface properties. Strategies to enhance EPR effect include tumor vasculature normalization (bevacizumab), mild hyperthermia (39–42°C for 30–60 min before nanoparticle administration), and pharmaceutical modulation of vascular permeability. Zwitterionic functionalization directly enhances EPR-based targeting by providing resistance to plasma protein adsorption and macrophage phagocytosis, thereby prolonging systemic circulation and increasing the probability of tumor accumulation across multiple vascular passages.
2.5 Comparave Nanoparticle Analysis
A comparative understanding of polymeric, lipidic, and inorganic nanoparticle systems provides essential context for appreciating the unique advantages of mesoporous silica nanoparticles.
Polymeric nanoparticles fabricated from PLGA, PLA, PCL, and chitosan offer sustained release through polymer matrix degradation, excellent encapsulation efficiency, and ability to incorporate targeting ligands. However, they suffer from batch-to-batch variability, relatively low drug loading capacity (~5–15%), and potential toxicity of accumulated polymer degradation products.
Liposomes are clinically most advanced, with Doxil and Abraxane benchmarks. However, liposomal systems suffer from physical instability during storage, drug leakage, opsonization-induced rapid clearance, and limited loading capacity for hydrophobic drugs unless incorporated into the bilayer.
Gold nanoparticles offer photothermal therapy potential but raise concerns regarding long-term biodistribution, hepatic/splenic accumulation, and clearance.
Iron oxide nanoparticles find application as MRI contrast agents and in magnetic hyperthermia but have limited drug loading capacity (~1–5%).
Mesoporous silica nanoparticles stand apart through their exceptionally high surface area (700–1200 m²/g), tunable pore diameter (2–10 nm), large pore volume (0.5–2.5 cm³/g), narrow pore size distribution enabling size-selective loading, and versatile silanol surface chemistry. MSNs can accoodate substantially higher drug loading (20–40% w/w) than other systems, and the silanol chemistry permits covalent modification with amino, carboxyl, thiol, epoxide, and zwitterionic groups through well-established silane chemistry.
3. BIOLOGICAL BARRIERS TO NANOPARTICLE DELIVERY AND PROTEIN CORONA
3.1 Systemic Circulation Barriers
The first barrier encountered by intravenously administered nanoparticles is blood itself. Plasma proteins including albumin (most abundant, 35–50 g/L), fibrinogen, immunoglobulins, complement proteins, and apolipoproteins rapidly adsorb onto nanoparticle surfaces forming a protein corona within seconds to minutes of exposure. This protein corona fundamentally alters the biological identity of the nanoparticle, masks surface targeting ligands, alters cellular uptake pathways, and triggers MPS recognition. Rapid MPS-mediated clearance in liver and spleen (splenic macrophages) dramatically reduces the fraction of administered nanoparticles reaching tumors, with estimates suggesting <1% of injected dose typically accumulating at tumor sites even under optimal conditions.
3.2 Protein Cona Formation and Composition
Protein corona formation is a dynamic two-stage process :
Stage 1: Abundant high-affinity proteins rapidly adsorb, forming a tightly associated monolayer with residence times >hours. The hard corona composition directly reflects the thermodynamic equilibrium between protein-nanoparticle affinity and protein-protein displacement dynamics.
Stage 2: Lower-affinity proteins form a more loosely associated secondary layer with rapid exchange kinetics (seconds to minutes).
Corona composition is dictated by surface charge (positive surfaces attract anionic proteins like albumin; negative surfaces selectively adsorb cationic proteins), hydrophobicity (hydrophobic surfaces attract fibrinogen, IgG), curvature (high-curvature small nanoparticles show different corona profile than larger), and functional groups. For positively charged or hydrophobic surfaces, corona formation is pronounced, leading to rapid opsonization.
The corona dictates biological fate in ways that can be either deleterious or beneficial. Opsonins (immunoglobulins, complement C3b, C4b, C-reactive protein) promote phagocytosis by tagging nanoparticles for MPS recognition. Dysopsonins (clusterin, apolipoproteins, certain albumin conformations) reduce macrophage uptake and may redirect nanoparticles to alternative biodistribution pathways. Corona-driven uptake may occasionally be useful (e.g., apolipoprotein E binding enables LDL-receptor-mediated uptake across the blood-brain barrier), but generally constitutes a major barrier to selective tumor targeting.
3.3 Zwitterion Coatings Versus PEG
PEGylation has been the conventional gold-standard approach for creating stealth nanoparticles, operating through steric repulsion against protein adsorption. However, PEG suffrs from critical limitations:
Zwitterionic functionalization addresses each limitation through a fundamentally different mechanism. The simultaneous present cationic and anionic groups of zwitterions create a strong electric dipole that electrostatically attracts and organizes water molecules into a tightly bound hydration layer. This hydration shell acts as a physical and energetic barrier—proteins approaching the surface must displace bound water, which is thermodynamically costly. Zwitterionic polymer brushes grafted onto planar surfaces and nanoparticles reduce nonspecific protein adsorption from undiluted blood plasma to <0.3 ng/cm², substantially below complement activation thresholds. Critically, zwitterionic coatings do not trigger anti-coating antibody formation upon repeated dosing, providing durable, repeatable stealth functionality suitable for sustained therapeutic regimens.
4. MESOPOROUS SILICA NANOPARTICLES: STRUCTURE, SYNTHESIS, AND SURFACE CHEMISTRY
4.1 Structural Architecture
MSNs possess regular arrangements of cylindrical mesopores (2–50 nm by IUPAC classification) creating enormous internal surface area for drug loading. The principal architectural types include:
MCM-41: Two-dimensional hexagonal arrangement of parallel cylindrical pores, pore size 2–5 nm, high surface area (~1000 m²/g), high pore volume (~1 cm³/g). Most extensively studied for drug delivery due to well-characterized hexagonal symmetry.
MCM-48: Three-dimensional cubic bicontinuous pore network, pore size 2–4 nm, surface area ~900 m²/g, provides improved diffusion/connectivity versus 2D systems.
SBA-15: Larger pore diameters (5–10 nm) with thicker pore walls (3–6 nm) and microporous interconnections, conferring greater hydrothermal and mechanical stability.
MSN surface silanol groups exist as isolated (single Si-OH on one silicon), geminal (two Si-OH on the same silicon), and vicinal (adjacent Si-OH on neighboring silicons connected through hydrogen bonding) forms at density ~4–5 OH/nm², providing a total of ~2-5 × 10¹⁸ silanol groups per gram of MSN for versatile functionalization.
5.2 Sol-Gel Synthesis
MSNs are synthesized through sol-gel condensation of silica precursors in the presence of structure-directing surfactants:
The synthesis proceeds through: surfactant dissolution and self-assembly into micelles above CMC (critical micelle concentration); silica precursor addition undergoing hydrolysis (Si-OR + H₂O → Si-OH + ROH) and condensation (Si-OH + Si-OH → Si-O-Si + H₂O or Si-OH + Si-OR → Si-O-Si + ROH); silica condensation around micellar templates; aging and strengthening of the silica framework; surfactant template removal by calcination (550°C for 5 h) or solvent extraction (HCl/ethanol reflux or NH₄NO₃/ethanol).
Particle size, pore diameter, pore volume, and morphology are controlled by adjusting silica-to-surfactant ratio, reaction temperature, pH, co-solvent addition (ethanol slows hydrolysis), and pore expanders (trimethylbenzene enlarges pore diameter for SBA-type). The Box-Behnken optimization in the present thesis exploits this tunability through systematic variation of CTAB amount (0.25–1.0 g), TEOS volume (5.0–10.0 mL), and reaction temperature (65–90°C).
4.3 Surface Functionalization
Two principal approaches enable surface modification of MSNs:
Post-synthesis grafting: Organosilane reagents react with preformed MSN silanols via hydrolysis/condensation. This offers precise control over functional group density, minimizes mesostructure disruption, and preserves drug-loading capability by permitting drug loading after functionalization. However, uneven distribution can occur with preferential external surface modification.
Co-condensation (one-pot): Simultaneous condensation of TEOS and organosilane during MSN synthesis yields homogeneous organic group distribution throughout the framework but risks mesostructure disruption if organosilane >10–20 mol% and cannot generally be applied after drug loading.
Common functionalizations include amines (APTES, providing –NH₂), carboxyls (carboxyethylsilanetriol), thiols, epoxides (3-glycidoxypropyltrimethoxysilane), and zwitterionic groups. The zwitterionic functionalization architecture employed in the present study involves three sequential steps: APTES-mediated amine grafting, deoxycholic acid conjugation via EDC/NHS coupling, and sulfobetaine 12 self-assembly.
5. ZWITTERIONIC MERIALS: CHEMISTRY, CLASSIFICATION, AND BIOLOGICAL RELEVANCE
5.1 Molecular Basis of Zwitterionic Hydration
Zwitterionic materials contain both cationic and anionic functional groups within the same molecular unit with overall electrical neutrality. The coexistence of opposite charges creates a strong electric dipole that interacts powerfully with surrounding water molecules through electrostatic mechanisms. The quaternary ammonium cation (–N⁺(CH₃)₃–) electrostatically attracts the oxygen atoms of water molecules, while the sulfonate (–SO₃⁻) or carboxylate (–COO⁻) anion attracts the hydrogen atoms of water. This dual coordination organizes water molecules into a tightly bound hydration layer with binding energies of 50–100 kJ/mol per water molecule, compared to ~20–30 kJ/mol for PEG-mediated hydrogen bonding hydration. The thermodynamic stability of this hydration layer exceeds that of conventional hydrophilic polymer hydration, providing the molecular foundation for exceptional antifouling.
5.2 Classification Zwitterionic Materials
Three principal zwitterionic families are employed in biomedical applications:
Phosphorylcholine-bas zwitterions are naturally occurring zwitterionic head groups found in the outer leaflet of mammalian cell membranes. PC-based polymers, including poly(2-methacryloyloxyethyl phosphorylcholine), mimic cell membrane non-fouling properties with excellent hemocompatibility and low immunogenicity. Applications include coronary stent coatings, contact lenses, and DNA delivery vectors.
Sulfobetaine-based zwitterions contain a quaternary ammonium cation and sulfonate anion in the same molecule, separated by a short alkyl spacer (typically propyl). Poly(sulfobetaine methacrylate) coatings exhibit outstanding antifouling properties with protein adsorption <0.3 ng/cm² from undiluted plasma. 3-(Dimethyl(3-(trimethoxysilylpropyl)ammonio)propane-1-sulfonate), known as zwitterionic silane or DMAPS-Si, enables direct grafting of sulfobetaine groups onto silica surfaces. The antifouling performance of PSBMA remains robust under high salt and elevated temperatures, conditions where PEG performance degrades.
Carboxybetaine-based zwitterions contain a quaternary ammonium cation and a carboxylate anion. The functional carboxyl group can be chemically activated via EDC/NHS coupling for conjugation of targeting ligands, peptides, antibodies, or other functional moieties without compromising zwitterionic character. This unique bifunctionality makes CB particularly attractive for targeted antifouling nanoparticle systems.
The sulfobetaine 12 (SB12: N-dodecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate) used in the present thesis is a small-molecule zwitterionic surfactant containing a long C12 hydrophobic tail and sulfobetaine head group, capable of self-assembly onto hydrophobic deoxycholic acid-modified MSN surfaces forming a stable zwitterionic outer shell.
5.3 Antifouling Mechanism and Quantification
The antifouling mechanism of zwitterionic coatings operates through a fundamentally different molecular mechanism compared to PEG-based steric repulsion. Zwitterionic coatings crate a tightly bound hydration shell through electrostatically induced water dipole alignment, with binding enthalpies of -50 to -80 kJ/mol per zwitterionic repeat unit as determined by surface plasmon resonance, isothermal titration calorimetry, and quartz crystal microbalance with dissipation measurements. Protein adsorption onto a zwitterionic brush-coated surface requires displacement of this bound water, which is thermodynamically unfavorable at ΔG > +20 kJ/mol for typical plasma proteins.
Quantitative antifouling performance: protein adsorption from undiluted human blood plasma has been measured at <0.3 ng/cm² for zwitterionic polymer brushes versus 5–50 ng/cm² for PEG brushes under identical conditions. Bacterial adhesion is reduced by >99% versus bare controls. Fibroblast cell adhesion onto zwitterionic-modified surfaces is reduced by >95% versus bare surfaces.
In vivo, zwitterionic functionalized nanoparticles demonstrate extended blood circulation half-lives (t₁/₂ often >24 h versus 3–6 h for unmodified MSNs), reduced hepatic/splenic accumulation, enhanced tumor accumulation through the EPR effect (2–5× enhancement versus unmodified), and lack of anti-zwitterion antibody formation upon repeated dosing.
6. OROXYLIN A: PHARMACOGICAL PROFILE AND THERAPEUTIC POTENTIAL
6.1 Natural Origin, Chemistry, and Physicochemical Properties
Oroxylin A (5,7-dihydroxy-6-methoxyflavone; CAS Registry Number 480-11-5; Molecular Formula C₁₆H₁₂O₅; Molecular Weight 284.26 g/mol) is a bioactive flavonoid isolated from the roots and stem bark of Oroxylum indicum and Scutellaria baicalensis (Chinese skullcap/Baikal skullcap). The compound exists as a pale yellow to yellowish crystalline powder, odorless, freely soluble in methanol, ethanol, DMSO, and acetone, sparingly soluble in chloroform, and practically insoluble in water (0.073 ± 0.004 mg/mL).
The Oroxylin A molecule contains:
The compound's BCS Class II classification (low solubility, high permeability) underscores the biopharmaceutical challenge that necessitates a rationally engineered nanocarrier system capable of protecting Oroxylin A from premature degradation, improving its solubilization, and directing it selectively toward lung tumor tissue.
6.2 Anticancer Pharmacological Activities
Oroxylin A demonstrates multimodal anticancer activity through multiple mechanistic pathways:
G2/M phase cell cycle arrest: Oroxylin A induces G2/M arrest in cancer cells through upregulation of p21 WAF1/CIP1 and p27 KIP1 CDK inhibitors, downregulation of cyclin B1 and Cdc2, and activation of ATM/ATR DNA damage response pathways.
Apoptosis induction: Oroxylin A activates both intrinsic (mitochondrial) and extrinsic (death receptor) apoptotic pathways. Intrinsic pathway activation involves downregulation of Bcl-2 and Bcl-xL, upregulation of Bax and Bak, mitochondrial membrane potential loss, cytochrome c release, caspase-9 and caspase-3 activation. Extrinsic pathway involves FasL upregulation and caspase-8 activation.
PI3K/AKT/mTOR pathway suppression: Oroxylin A inhibits AKT phosphorylation at Ser473, suppressing downstream mTORC1 activation, reducing phosphorylation of S6K1 and 4E-BP1, and inhibiting oncogenic cap-dependent translation. This suppresses tumor growth, survival, and proliferation signals.
NF-κB pathway inhibition: Oroxylin A prevents IκBα phosphorylation and degradation, retaining NF-κB in the cytoplasm and preventing its nuclear translocation and anti-apoptotic gene transcription. This sensitizes cancer cells to apoptotic stimuli.
Warburg effect suppression: Oroxylin A inhibits aerobic glycolysis in cancer cells through downregulation of GLUT1, HK2, PKM2, and LDHA, redirecting energy metabolism away from glycolysis and reducing lactate production, with potential normalization of tumor microenvironment pH.
MDM2 downregulation and p53 activation: Oroxylin A disrupts the MDM2-p53 negative feedback loop by downregulating MDM2 expression, leading to p53 stabilization, nuclear accumulation, and activation of p53 target genes for cell cycle arrest and apoptosis.
Tumor invasion and metastasis suppression: Oroxylin A inhibits MMP-2 and MMP-9 expression and enzymatic activity through suppression of NF-κB-mediated transcriptional activation, reducing ECM degradation and metastatic potential. Inhibition of EMT transcription factors further reduces metastatic capacity.
6.3 Pharmacokinetics and Biopharmaceutical Challenges
Despite this compelling pharmacological profile, clinical translation of Oroxylin A is severely hampered by fundamental biopharmaceutical limitations:
These interrelated limitations collectively establish the rationale for the zwitterionic MSN delivery platform developed in the present thesis, providing solubilization (mesopore loading transforms crystalline Oroxylin A into amorphous form), protection, passive targeting (zwitterionic stealth functionality prolongs circulation), and enhanced cellular uptake (zwitterionic dipole-mediated interactions increase endocytosis).
7. RATIONALE FOR ZWITTERIONIC-MSN DELIVERY SYSTEM
The integration of mesoporous silica nanoparticles and zwitterionic coatings in the Oroxylin A delivery platform addresses multiple interdependent therapeutic challenges:
The design strategy employs a three-component surface architecture:
The selection of SB12 specifically over carboxybetaine alternatives was motivated by: (i) SB12's commercial availability as a long-tail amphiphilic zwitterion providing simple self-assembly on hydrophobic surfaces; (ii) the well-characterized MTT assay performance of SB12-coated systems in cancer models; (iii) the demonstrated precedent of Gao et al. using DC@SB12-modified MSNs for protein drug oral delivery, validating the chemistry for the present application.
8. PREFORMULATION CHARACTERIZATION OF OROXYLIN A
8.1 Organoleptic Properties and Identification
Oroxylin A was visually examined and the observed characteristics were found to be in close agreement with reported literature values. The drug appeared as a yellow crystalline powder, consistent with flavonoid-class chemistry and the conjugated chromophore responsible for characteristic yellow coloration. It was odourless and existed as a solid at room temperature. These organoleptic properties align precisely with reported values, confirming identity and integrity of the procured drug sample, validating its suitability for subsequent preformulation and formulation studies.
Table 1: Organoleptic Properties and Identification of Oroxylin A
|
Parameter |
Reported Value |
Observed Value |
|
Appearance |
Crystalline powder |
Crystalline powder |
|
Colour |
Yellow |
Yellow |
|
Odour |
Odourless |
Odourless |
|
Nature |
Solid |
Solid |
8.2 Melting Point and Thermal Behavior
Melting point determination using the open capillary tube method showed 196–198°C, in close agreement with the reported literature value of 195–197°C. This narrow 2°C experimental deviation confirms the identity and high purity of the drug substance. DSC analysis of pure Oroxylin A revealed a sharp endothermic melting peak at 196.8°C, confirming crystalline nature and indicating minimal impurities.
8.3 UV Spectrophotometric Analysis and Calibration
UV scanning in methanol over 200–400 nm revealed dual absorption maxima at 273 nm and 321 nm, attributed to π→π* and n→π* electronic transitions of the flavone chromophore system. The 273 nm wavelength was selected as the analytical detection wavelength for all subsequent quantitative determinations due to higher sensitivity and minimal interference from potential excipient UV absorption.
A calibration curve constructed at 273 nm over 2–12 µg/mL demonstrated excellent linearity with the regression equation y = 0.0554x + 0.0033 and R² = 0.9995, confirming strict Beer-Lambert compliance.
Table 2: Calibration Curve Data of Oroxylin A in Methanol at 273 nm
|
Concentration (µg/mL) |
Absorbance |
|
2 |
0.124 ± 0.003 |
|
4 |
0.221 ± 0.005 |
|
6 |
0.334 ± 0.004 |
|
8 |
0.441 ± 0.006 |
|
10 |
0.558 ± 0.007 |
|
12 |
0.671 ± 0.005 |
Values expressed as mean ± SD (n = 3). The strongly linear calibration curve enables accurate quantification of Oroxylin A in entrapment efficiency, drug loading, and in vitro drug release studies.
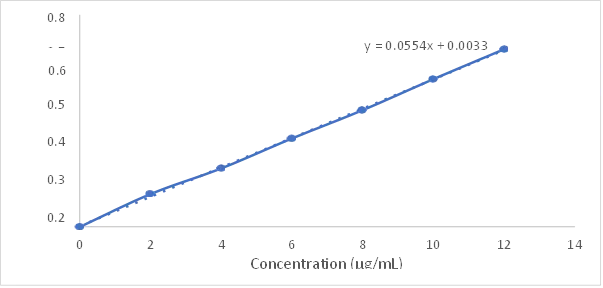
Fig 1: Calibration curve of Oroxylin A in methanol at 273 nm showing linearity across 2–12 µg/mL
8.4 Solubility Assessment
Saturation shake-flask analysis at 25 ± 0.5°C over 24 hours indicated:
Table 3: Solubility of Oroxylin A
|
Solvent |
Observed Solubility (mg/mL) |
Classification |
|
Distilled Water |
0.073 ± 0.004 |
Practically Insoluble |
|
Phosphate Buffer pH 7.4 |
0.087 ± 0.003 |
Practically Insoluble |
|
Phosphate Buffer pH 6.8 |
0.069 ± 0.005 |
Practically Insoluble |
|
Ethanol |
8.34 ± 0.21 |
Slightly Soluble |
|
Methanol |
11.62 ± 0.34 |
Sparingly Soluble |
|
DMSO |
61.43 ± 0.87 |
Freely Soluble |
|
Acetone |
4.17 ± 0.18 |
Slightly Soluble |
|
Chloroform |
6.82 ± 0.29 |
Slightly Soluble |
These data confirm Oroxylin A is BCS Class II (low solubility, high permeability) and rationalize the use of methanol (optimal balance of solubilization and volatility for rotary evaporation loading) as the loading solvent, while underscoring the necessity of nanoparticulate formulation.
8.5 FTIR Identification
Fourier transform infrared (FTIR) spectroscopy confirmed the successful synthesis, surface modification, and drug encapsulation within the mesoporous silica nanoparticle system. Pure Oroxylin A exhibited characteristic absorption bands corresponding to phenolic hydroxyl groups, flavone carbonyl functionality, and aromatic ring vibrations, verifying the structural integrity of the drug molecule. Bare mesoporous silica nanoparticles displayed the characteristic silanol and siloxane stretching vibrations associated with the silica framework.
Fourier transform infrared (FTIR) spectroscopy confirmed the successful synthesis, surface modification, and drug encapsulation within the mesoporous silica nanoparticle system. Pure Oroxylin A exhibited characteristic absorption bands corresponding to phenolic hydroxyl groups, flavone carbonyl functionality, and aromatic ring vibrations, verifying the structural integrity of the drug molecule. Bare mesoporous silica nanoparticles displayed the characteristic silanol and siloxane stretching vibrations associated with the silica framework.
The FTIR analysis demonstrated the successful fabrication of zwitterionic functionalized mesoporous silica nanoparticles capable of efficiently incorporating Oroxylin A while maintaining the chemical integrity of both the drug and the nanocarrier components.
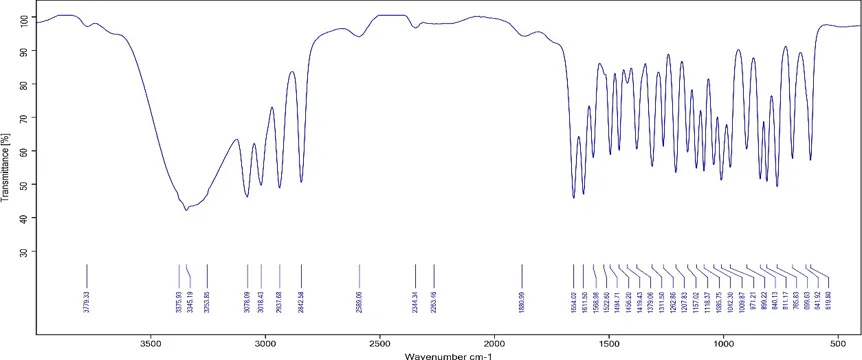
Fig 2: FTIR spectrum of Pure Oroxylin A
9. SYNTHESIS AND SEQUENTIAL FUNCTIONALIZATION PROTOCOL
9.1 MSN Core Synthesis
Bare MSNs were prepared by surfactant-templated sol-gel method. CTAB (0.25–1.00 g, per BBD design) was dissolved in distilled water under stirring, pH adjusted with sodium hydroxide solution (NaOH catalysis ~0.3 M), the mixture heated to 65–90°C (per design), and TEOS (5.0–10.0 mL, per design) added dropwise to initiate hydrolysis and condensation around CTAB micelles. After 4 h reaction, the white precipitate was centrifuged at 10,000 rpm for 10 min, washed thrice with distilled water and ethanol, dried overnight at 80°C, and calcined at 550°C for 5 h in a muffle furnace to remove surfactant, yielding bare MSNs with well-ordered cylindrical mesopores.
9.2 Three-Step Sequent Zwitterionic Functionalization
A sequential three-step post-synthetic surface modification protocol was employed for OXA-loaded MSNs, with each step introduced through precise chemical reactivity:
Step 1: OXA-MSN (100 mg) was dispersed in 40 mL anhydrous ethanol under probe sonication (10 min for homogeneous dispersion), 0.8 mL APTES added dropwise under nitrogen atmosphere to prevent oxidation of the amine, the mixture refluxed at 80°C for 18 h, yielding OXA-MSN-NH₂ via covalent siloxane bond formation between APTES triethoxysilyl groups and MSN surface silanols, releasing ethanol as byproduct. Recovery involved centrifugation at 10,000 rpm for 10 min, washing thrice with anhydrous ethanol to remove unreacted APTES, and vacuum drying at 50°C.
Step 2: OXA-MSN-NH₂ (100 mg was dispersed in 50 mL DMSO, to which 100 mg deoxycholic acid, 50 mg N-hydroxysuccinimide, and 50 mg EDC·HCl (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride) were sequentially added. The mixture reacted at 60°C for 48 h. The EDC/NHS-mediated amide coupling proceeds through: (i) EDC activation of DCA's carboxyl group forming O-acylisourea intermediate; (ii) NHS displacement forming more stable NHS-ester; (iii) amine nucleophilic attack forming covalent amide bond (–CO-NH–) with EDC/NHS byproduct removal. This yields OXA-MSN-DC with hydrophobic DCA inner coating presenting carboxylate groups available for further functionalization. Recovery involved centrifugation, washing with DMSO then distilled water, overnight drying.
Step 3 (SB12 Zwitterionic Sf-Assembly): OXA-MSN-DC (50 mg) was dispersed in 10 mL distilled water under sonication, a solution of 50 mg SB12 in 5 mL distilled water added dropwise at 500 rpm for 60 min at room temperature, during which SB12 (N-dodecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate) self-assembled onto the DCA-modified surface through hydrophobic C12 tail interactions with DCA steroid rings and electrostatic interactions between SB12 sulfonate and residual surface cationic groups. This generated a net-zero charge zwitterionic outer shell bearing both quaternary ammonium (–N⁺(CH₃)₂–) and sulfonate (–SO₃⁻) groups. The final OXA-SB12-MSN was recovered by centrifugation at 12,000 rpm for 10 min, washed thrice with distilled water, lyophilized, and stored at 4°C protected from light.
9.3.. Box-Behnken Design-Dri Formulation Optimization
9.3.1 Methodology Rationale
Three independent variables critical to MSN physicochemical properties were selected based on preliminary screening and literature evidence:
A three-factor, three-level Box-Behnken Design using Design Expert® 13 generated 17 experimental runs (5 center points for reproducibility assessment, 8 factorial runs, 4 axial runs) with particle size (Y₁, minimize) and entrapment efficiency (Y₂, maximize) as dependent responses modeled via quadratic polynomial equations.
9.3.2 ANOVA for Particle Size (Y₁)
The quadratic model for particle size demonstrated excellent statistical significance (F = 76.75; p < 0.0001), indicating the model explained the variance in particle size at >99.99% confidence level. Lack of fit was non-significant F = 0.9570, p = 0.4940), confirming model adequacy in representing experimental data without systematic deviation. CTAB amount exerted the most dominant linear influence (F = 434.00; p < 0.0001), followed by TEOS volume (F = 163.93; p < 0.0001), while reaction temperature alone was non-significant (F = 0.1515, p = 0.7087). The interaction term BC (TEOS × Temperature) was significant (p = 0.0367), indicating non-additive effects of these variables. Quadratic terms A² (p = 0.0405), B² (p = 0.0003), and C² (p = 0.0024) were significant, confirming non-linear response surfaces. Fit statistics indicated excellent model reliability: R² = 0.9900, adjusted R² = 0.9771, predicted R² = 0.9238, with less than 0.2 difference between adjusted and predicted R² confirming strong predictive capability. The adequate precision value of 31.01 indicated strong signal-to-noise ratio.
The regression equation was:
Y₁ = 163.52 − 50.175A + 30.8375B − 0.9375C + 6.35AB + 5.6AC + 8.775BC + 8.3275A² + 22.3025B² + 15.3525C²
The dominant negative coefficient of A (−50.175) confirmed that increasing CTAB substantially reduces particle size through denser micellar templating, while the positive coefficient of B (+30.8375) confirmed that increasing TEOS progressively increases particle size through enhanced silica condensation and particle growth.
Table 4 : ANOVA for the quadratic model for Particle Size (Y1)
|
Source |
Sum of Squares |
df |
Mean Square |
F-value |
p-value |
|
|
Model |
32056.92 |
9 |
3561.88 |
76.75 |
< 0.0001 |
significant |
|
A-CTAB |
20140.24 |
1 |
20140.24 |
434.00 |
< 0.0001 |
|
|
B-TEOS |
7607.61 |
1 |
7607.61 |
163.93 |
< 0.0001 |
|
|
C-Reaction Temperature |
7.03 |
1 |
7.03 |
0.1515 |
0.7087 |
|
|
AB |
161.29 |
1 |
161.29 |
3.48 |
0.1046 |
|
|
AC |
125.44 |
1 |
125.44 |
2.70 |
0.1442 |
|
|
BC |
308.00 |
1 |
308.00 |
6.64 |
0.0367 |
|
|
A² |
291.99 |
1 |
291.99 |
6.29 |
0.0405 |
|
|
B² |
2094.32 |
1 |
2094.32 |
45.13 |
0.0003 |
|
|
C² |
992.42 |
1 |
992.42 |
21.39 |
0.0024 |
|
|
Residual |
324.85 |
7 |
46.41 |
|
|
|
|
Lack of Fit |
135.74 |
3 |
45.25 |
0.9570 |
0.4940 |
Not significant |
|
Pure Error |
189.11 |
4 |
47.28 |
|
|
|
|
Cor Total |
32381.76 |
16 |
|
|
|
|
9.3.3. ANOVA for Entrapment Efficiency (Y₂)
The quadratic model for entrapment efficiency was statistically significant (F = 19.21; p = 0.0004). Lack of fit was non-significant (F = 0.5159, p = 0.6933). CTAB amount (F = 106.37; p < 0.0001) was the most influential variable, while TEOS volume exerted a significant negative influence (F = 28.27; p = 0.0011). The interaction term AB was significant (p = 0.0483), and quadratic terms B² (p = 0.0055) and C² (p = 0.0250) were significant. R² = 0.9611 and adjusted R² = 0.9110 indicated good model fit; adequate precision of 14.41 confirmed a satisfactory signal-to-noise ratio.
The regression equation was:
Y₂ = 77.14 + 9.2375A − 4.7625B + 0.4C − 3.025AB − 1.15AC − 0.55BC − 2.6325A² − 4.8825B² − 3.5075C²
Higher CTAB levels combined with lower TEOS volumes generated well-ordered mesopore channels with greater surface area, facilitating enhanced drug encapsulation. The negative B coefficient (−4.7625) confirmed that excess TEOS leads to denser silica walls with reduced accessible pore volume.
Table 5 : ANOVA for the quadratic model for Entrapment Efficiency (Y2)
|
Source |
Sum of Squares |
df |
Mean Square |
F-value |
p-value |
|
|
Model |
1109.36 |
9 |
123.26 |
19.21 |
0.0004 |
significant |
|
A-CTAB |
682.65 |
1 |
682.65 |
106.37 |
< 0.0001 |
|
|
B-TEOS |
181.45 |
1 |
181.45 |
28.27 |
0.0011 |
|
|
C-Reaction Temperature |
1.28 |
1 |
1.28 |
0.1994 |
0.6687 |
|
|
AB |
36.60 |
1 |
36.60 |
5.70 |
0.0483 |
|
|
AC |
5.29 |
1 |
5.29 |
0.8243 |
0.3941 |
|
|
BC |
1.21 |
1 |
1.21 |
0.1885 |
0.6772 |
|
|
A² |
29.18 |
1 |
29.18 |
4.55 |
0.0704 |
|
|
B² |
100.37 |
1 |
100.37 |
15.64 |
0.0055 |
|
|
C² |
51.80 |
1 |
51.80 |
8.07 |
0.0250 |
|
|
Residual |
44.92 |
7 |
6.42 |
|
|
|
|
Lack of Fit |
12.53 |
3 |
4.18 |
0.5159 |
0.6933 |
Not significant |
|
Pure Error |
32.39 |
4 |
8.10 |
|
|
|
|
Cor Total |
1154.28 |
16 |
|
|
|
|
Table 6: Fit statistics of Entrapment Efficiency (Y2)
|
Std. Dev. |
2.53 |
R² |
0.9611 |
|
Mean |
71.95 |
Adjusted R² |
0.9110 |
|
C.V. % |
3.52 |
Predicted R² |
0.7824 |
|
|
|
Adeq Precision |
14.4109 |
9.3.4.Physicochemical Responses Across Batches
Across the 17 BBD batches:
Table 7: Physicochemical Evaluation Data of Oroxylin A–Zwitterionic Mesoporous Silica Nanoparticle Formulation Batches (RF1–RF17)
|
Batch |
Particle Size (nm) PDI |
Zeta Potential (mV) |
% EE |
% DL |
% Yield |
|
|
RF1 |
155.3 ± 4.8 |
0.212 ± 0.018 |
−18.2 ± 1.4 |
73.8 ± 1.8 |
19.6 ± 0.9 |
81.4 ± 1.4 |
|
RF2 |
171.4 ± 5.6 |
0.234 ± 0.021 |
−19.8 ± 1.7 |
80.4 ± 2.1 |
21.2 ± 1.1 |
85.2 ± 1.6 |
|
RF3 |
218.3 ± 4.6 |
0.288 ± 0.018 |
−22.4 ± 1.3 |
63.4 ± 1.8 |
17.4 ± 0.8 |
76.4 ± 1.3 |
|
RF4 |
241.7 ± 5.2 |
0.314 ± 0.021 |
−22.8 ± 1.5 |
65.2 ± 2.1 |
17.8 ± 0.9 |
74.8 ± 1.4 |
|
RF5 |
128.6 ± 3.1 |
0.184 ± 0.011 |
−16.4 ± 0.9 |
82.6 ± 1.6 |
21.5 ± 0.7 |
85.4 ± 1.0 |
|
RF6 |
112.4 ± 2.7 |
0.162 ± 0.008 |
−12.7 ± 1.2 |
86.4 ± 1.3 |
22.3 ± 0.6 |
88.6 ± 0.8 |
|
RF7 |
178.2 ± 3.8 |
0.256 ± 0.014 |
−17.8 ± 1.1 |
71.2 ± 1.5 |
19.1 ± 0.8 |
79.2 ± 1.2 |
|
RF8 |
158.4 ± 3.4 |
0.231 ± 0.013 |
−15.3 ± 0.7 |
74.8 ± 1.7 |
19.9 ± 0.6 |
81.6 ± 1.1 |
|
RF9 |
234.6 ± 5.8 |
0.328 ± 0.019 |
−26.4 ± 1.6 |
61.7 ± 2.3 |
17.0 ± 1.1 |
73.8 ± 1.6 |
|
RF10 |
138.3 ± 3.2 |
0.196 ± 0.012 |
−14.6 ± 1.0 |
79.4 ± 1.8 |
20.8 ± 0.8 |
84.2 ± 1.0 |
|
RF11 |
163.6 ± 5.2 |
0.226 ± 0.019 |
−19.1 ± 1.6 |
77.3 ± 2.0 |
20.4 ± 1.0 |
83.1 ± 1.5 |
|
RF12 |
169.1 ± 6.1 |
0.231 ± 0.022 |
−19.6 ± 1.8 |
79.4 ± 2.3 |
21.0 ± 1.2 |
84.3 ± 1.7 |
|
RF13 |
263.2 ± 6.4 |
0.381 ± 0.024 |
−31.6 ± 1.8 |
58.9 ± 2.6 |
16.3 ± 1.2 |
72.4 ± 1.7 |
|
RF14 |
182.7 ± 4.1 |
0.267 ± 0.016 |
−16.8 ± 1.1 |
69.8 ± 1.9 |
18.8 ± 0.9 |
78.6 ± 1.3 |
|
RF15 |
226.4 ± 5.1 |
0.318 ± 0.020 |
−21.4 ± 1.4 |
63.8 ± 2.0 |
17.5 ± 0.8 |
75.2 ± 1.4 |
|
RF16 |
247.3 ± 5.7 |
0.356 ± 0.022 |
−28.4 ± 1.7 |
60.3 ± 2.4 |
16.6 ± 1.0 |
73.6 ± 1.5 |
|
RF17 |
158.2 ± 4.4 |
0.218 ± 0.017 |
−18.4 ± 1.5 |
74.8 ± 1.9 |
19.9 ± 0.9 |
82.2 ± 1.4 |
n = 3; SD = Standard Deviation; EE = Entrapment Efficiency; DL = Drug Loading.
Higher CTAB concentrations promoted smaller, more uniform particles through denser micelle templating, while increased TEOS volume led to larger aggregated particles via excessive silica condensation. PDI was narrower in batches with higher CTAB and lower TEOS, indicating improved homogeneity. Zeta potentials were negative across all batches reflecting partial silanol deprotonation (pKa ~6.5–7.0) compensated by SB12 zwitterionic groups. Entrapment efficiency directly correlated with particle size and available mesopore surface area.
9.3.5. Optimization and Validation
Desirability function optimization (Design Expert® 13) identified the optimal formulation at CTAB 1.0 g, TEOS 5.0 mL, and reaction temperature 77.5°C. Predicted particle size was 108.20 nm and entrapment efficiency 86.40%, with overall desirability of 0.9956—indicating excellent proximity to the theoretical optimum. Experimentally obtained values of 112.4 ± 2.7 nm and 86.4 ± 1.3% showed excellent agreement with percentage biases of only 3.88% and 0.00%, respectively, confirming the robustness and predictive accuracy of the quadratic models.
9.4Characterization of the Optimized Formulation
9.4.1 Particle Size, PDI, and Zeta Potential
Dynamic light scattering revealed particle size of 112.4 ± 2.7 nm with PDI of 0.162 ± 0.008, indicating narrow monomodal size distribution suitable for EPR-mediated tumor accumulation and clathrin-mediated endocytic uptake (optimal 60–200 nm range). The PDI <0.2 indicates good monodispersity important for reproducible in vivo behavior and consistent clinical performance. Zeta potential of −12.7 ± 1.2 mV reflects partial silanol deprotonation compensated by neutralization through net-zero charge zwitterionic SB12 groups, ensuring adequate colloidal stabilization under physiological conditions while reducing non-specific electrostatic interactions with healthy cell membranes.
9.4.2 FTIR Confirmation of Sequential Functionalization
Comparative FTIR analysis across pure Oroxylin A, bare MSNs, zwitterionic functionalized MSNs (OXA-SB12-MSN-DC), and drug-loaded MSNs (OXA-SB12-MSN) confirmed the sequential surface transformation:
Table 8: FTIR Spectral Interpretation of Pure Oroxylin A, Bare MSNs, Zwitterionic Functionalized MSNs, and Drug-Loaded MSNs
|
Assignment |
Pure Oroxylin A |
Bare MSNs |
OXA-SB12-MSN-DC |
Drug-Loaded OXA-SB12-MSN |
|
O–H stretching (phenolic/silanol) |
3779.33, 3375.93 |
3455.39 |
3445.51, 3416.14 |
3503.14, 3436.62 |
|
N–H stretching (amine/amide) |
3345.19 |
- |
3335.99 |
3245.29 |
|
Aromatic/ aliphatic C–H |
3078.09, 3018.43, 2937.68 |
2872.14 |
2925.18, 2853.97 |
3081.99, 3022.86, 2976.53, 2928.65, 2846.80 |
|
C=O (flavone/amide I) |
1654.02, 1611.50 |
1672.65 |
1642.78 |
1645.33, 1611.95 |
|
N–H bending (amide II) / C=C aromatic |
1568.98, 1522.60 |
- |
1564.29, 1544.87 |
1567.57, 1545.79 |
|
C–C aromatic ring stretching |
1494.71, 1456.20, 1419.43 |
1473.03 |
1480.57, 1455.68 |
1493.17, 1479.27, 1455.94 |
|
C–H bending / S=O symmetric |
1379.06, 1311.50 |
1301.21 |
1377.60, 1300.80 |
1377.44, 1304.89 |
|
C–O–C / S=O asymmetric |
1262.86, 1207.83, 1157.02 |
1136.19 |
1241.42, 1182.10 |
1262.34, 1239.86, 1208.96 |
|
Si–O–Si asymmetric stretching |
1118.37, 1085.75 |
1079.86 |
1116.40, 1080.18 |
1155.78, 1118.20, 1082.74 |
|
Si–OH / C–H out-of-plane bending |
971.21, 899.22, 840.13 |
960.20, 845.53, 800.89 |
1011.12, 873.03, 838.84 |
1010.09, 972.03, 897.32, 840.12 |
The emergence of N–H stretching at 3335 cm⁻¹ and amide I carbonyl at 1642 cm⁻¹ and amide II band at 1544 cm⁻¹ in OXA-SB12-MSN-DC confirmed successful APTES grafting and DCA conjugation via EDC/NHS-mediated amide bond formation. S=O stretching vibrations around 1300 and 1182 cm⁻¹ confirmed SB12 zwitterionic coating. The drug-loaded OXA-SB12-MSN retaed all functionalization peaks while displaying reappearance of Oroxylin A-specific aromatic C–H stretching at 3081 cm⁻¹ and C=O vibration at 1645 cm⁻¹, with notable broadening and intensity attenuation confirming molecular-level encapsulation and drug-carrier interaction.
9.4.3 DSC Thermogram
DSC thermogram of pure Oroxylin A exhibited a sharp endothermic melting peak at 196.8°C confirming crystalline nature and purity. The physical mixture showed retain drug melting transition at 198.2°C and a second event at 312.6°C attributed to the silicate framework thermal decomposition. Confirmation of physicochemical compatibility was evidenced by absence of new thermal events or significant peak shifts indicating no chemical interaction between Oroxylin A and the zwitterionic MSN carrier components.
9.4.4 Drug Loading Metrics
The drug-to-carrier ratio of 1:2 (w/w) optimized during preliminary screening was justified by considerations of mesopore saturation capacity, surface functionalization stability, and colloidal dispersibility. This ratio achieved drug loading of 22.3 ± 0.6% (DL%, calculated as mass of drug in nanoparticles × 100 / total mass of nanoparticles) and entrapment efficiency of 86.4 ± 1.3% (EE%, calculated as mass of drug entrapped × 100 / mass of drug added) in RF6, confirming high-capacity loading of hydrophobic Oroxylin A within the mesopore network.
9.5 In Vitro Drug Release and Kinetic Modeling
9.5.1 Biphasic Release Pattern
Cumulative in vitro drug release studies at pH 7.4 over 12 h using dialysis membrane diffusion method (12,000 Da MWCO, 50 mL PBS, 100 rpm, 37 ± 0.5°C) revealed clear concentration-dependent patterns governed primarily by particle size. All batches exhibited biphasic release:
RF6 (smallest particle, 112.4 nm) achieved the highest cumulative release of 76.8 ± 1.54% at 12 h, while RF13 (largest particle, 263.2 nm) released only 46.8 ± 0.98%. This clear inverse relationship between particle size and cumulative release reflects the diffusion path length through mesopore channels being shorter for smaller particles.
9.5.2 Kinetic Model Fitting
Drug release data for RF6 fitted five mathematical kinetic models:
Table 9: Kinetic Model Fitting
|
Kinetic Model |
R² |
Mechanism Indication |
|
Zero-order |
0.9766 |
Concentration-independent release (ideal for sustained delivery) |
|
First-order |
0.9952 |
Concentration-dependent release from reservoir |
|
Higuchi |
0.9805 |
Fickian diffusion from matrix |
|
Hixson-Crowell |
0.9981 |
Drug release from eroding matrix with surface area decreasing over time |
|
Korsmeyer-Peppas |
0.9988 |
Anomalous diffusion-controlled release |
The Korsmeyer-Peppas model provided the best fit with R² = 0.9988 and diffusional exponent n = 0.7250. The n value between 0.5 and 1.0 indicates anomalous non-Fickian diffusion transport, suggesting that Oroxylin A release from the zwitterionic MSN is governed by combined diffusion through mesopore channels AND erosion/relaxation of the surface functionalization layer. This dual release mechanism is therapeutically advantageous: initial burst provides rapid therapeutic onset, while sustained release maintains drug concentrations above cytotoxic thresholds over extended periods.
9.5.3 Physicochemical Drivers of Release Behavior
The biphasic release pattern is mechanistically driven by:
9.6. Accelerated Stability Under ICH Conditions
9.6.1 ICH-Compliant Protocol
The optimized OXA-SB12-MSN formulation was subjected to accelerated stability testing at 40 ± 2°C and 75 ± 5% RH for three months per ICH Q1A(R2) guidelines. Samples were evaluated at 0, 1, 2, and 3 months for:
9.6.2 Stability Findings
Across the three-month accelerated study, the formulation maintained excellent physicochemical stability:
Table 10: Stability study Oroxylin A
|
Parameter |
0 Month |
1 Month |
2 Month |
3 Month |
|
Appearance |
Yellow free-flowing powder |
Yellow free-flowing powder |
Yellow free-flowing powder |
Yellow free-flowing powder |
|
Particle Size (nm) |
112.4 ± 2.7 |
113.1 ± 2.9 |
113.8 ± 3.1 |
114.6 ± 3.2 |
|
PDI |
0.162 ± 0.008 |
0.164 ± 0.009 |
0.167 ± 0.010 |
0.169 ± 0.011 |
|
Zeta Potential (mV) |
−12.7 ± 1.2 |
−12.4 ± 1.3 |
−12.1 ± 1.4 |
−11.8 ± 1.4 |
|
% EE |
86.4 ± 1.3 |
85.9 ± 1.4 |
85.4 ± 1.5 |
84.8 ± 1.6 |
|
% CDR at 12 h |
76.8 ± 1.54 |
76.2 ± 1.58 |
75.6 ± 1.62 |
74.9 ± 1.68 |
The marginal variations across all parameters (≤2% particle size increase, ≤1.6% EE decrease, ≤1.9% CDR decrease) confirm excellent physicochemical stability under accelerated storage conditions, validating the formulation's potential for pharmaceutical development with adequate shelf-life. The slow rate of degradation suggests the formulation may also meet long-term stability requirements (25°C/60% RH), though this requires explicit verification at long-term storage conditions per ICH protocols.
9.6.3 Mechanistic Basis for Stability
The exceptional stability derives from the covalent siloxane anchoring of the APTES layer, the stable amide bond of DCA conjugation (amide hydrolysis negligible below 60°C), and the electrostatic/hydrophobic interactions stabilizing SB12 self-assembly. The minimal particle size change suggests absence of Ostwald ripening or aggregation during storage. Stable entrapment efficiency indicates absence of drug desorption or chemical degradation over the storage period.
9.7. In Vitro Cytotoxicity Against A549 Lung Cancer Cells
9.7.1 MTT Assay Methodology
Cytotoxicity was assessed against A549 human lung adenocarcinoma cells using the MTT assay at five concentrations (5, 10, 20, 40, 80 µg/mL) with 48 h treatment. A549 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C in 5% CO₂ humidified atmosphere. Cells were seeded in 96-well plates at 5×10³ cells/well and incubated 24 h for adherence before treatment. After 48 h exposure, 20 µL MTT (5 mg/mL) was added per well, 4 h incubation generated formazan crystals through mitochondrial dehydrogenase-mediated reduction of yellow MTT tetrazolium, crystals dissolved in 100 µL DMSO, and absorbance measured at 570 nm. Cell viability was calculated as:
Cell Viability (%) = (Absorbance_treated / Absorbance_control) × 100
IC₅₀ values were determined by non-linear regression analysis using GraphPad Prism software.
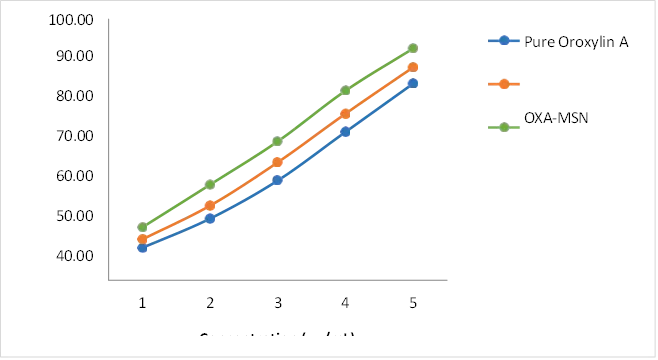
Fig 3:Concentration-dependent cytotoxicity (%)
9.7.2 Concentration-Dependent Cytotoxicity
Pure Oroxylin A exhibited IC₅₀ of 38.24 ± 1.46 µg/mL. Unfunctionalized OXA-MSN demonstrated inhanced cytotoxicity with IC₅₀ of 26.18 ± 1.22 µg/mL, reflecting improved drug delivery via mesoporous silica carrier. The zwitterionic-functionalized OXA-SB12-MSN-DC exhibited highest cytotoxic potency with IC₅₀ of 17.86 ± 0.94 µg/mL, representing a 2.14-fold improvement over pure Oroxylin A.
9.7.3 Comparative IC₅₀ Analysis
Table 11 : Comparative IC₅₀ Analysis
|
Sample |
IC₅₀ (µg/mL) |
Fold Reduction vs. Pure Drug |
|
Pure Oroxylin A |
38.24 ± 1.46 |
1.00 |
|
OXA-MSN (unfunctionalized) |
26.18 ± 1.22 |
1.46 |
|
OXA-SB12-MSN-DC (zwitterionic) |
17.86 ± 0.94 |
2.14 |
At 80 µg/mL concentration:
9.7.4 Mechanistic Interpretation
The superior cytotoxicity of OXA-SB12-MSN-DC arises from synergistic contributions:
9.7.5 Phase Contrast Microscopy Analysis
Microscopic examination at 48 h post-treatment revealed:
These morphological changes collectively suggest caspase-3 activation, DNA fragmentation, and apoptotic body formation at 48 h post-treatment with zwitterionic-functionalized nanoparticles.
10.CONCLUSION
Lung cancer continues to represent one of the greatest challenges in modern oncology owing to its high mortality rate, molecular heterogeneity, late-stage diagnosis, and the development of resistance to conventional therapeutic modalities. Although advances in targeted therapy and immunotherapy have improved clinical outcomes in selected patient populations, significant limitations related to systemic toxicity, poor tumor selectivity, multidrug resistance, and unfavorable pharmacokinetic profiles remain major barriers to effective treatment.
Nanotechnology-based drug delivery systems have emerged as promising strategies to overcome these challenges by improving drug solubility, biodistribution, tumor accumulation, and controlled release characteristics. Among the available nanocarriers, mesoporous silica nanoparticles have attracted considerable attention due to their large surface area, tunable pore structure, high drug-loading capacity, excellent biocompatibility, and versatile surface functionalization capabilities. Furthermore, zwitterionic surface engineering has provided an effective alternative to conventional stealth technologies by minimizing protein corona formation, reducing macrophage-mediated clearance, prolonging systemic circulation, and enhancing tumor accumulation through the enhanced permeability and retention effect.
Oroxylin A is a naturally occurring flavonoid with significant anticancer potential mediated through modulation of multiple oncogenic pathways, including PI3K/AKT/mTOR, NF-κB, apoptosis-related signaling, and tumor metabolic pathways. However, its poor aqueous solubility, rapid metabolism, short biological half-life, and limited bioavailability substantially restrict its clinical translation. The integration of Oroxylin A with zwitterionic functionalized mesoporous silica nanoparticles offers a rational and multifunctional therapeutic platform capable of addressing these limitations through improved drug protection, enhanced intracellular delivery, prolonged circulation, and sustained release behavior.
Collectively, the convergence of mesoporous silica nanotechnology, zwitterionic surface modification, and natural-product-based therapeutics represents a promising strategy for next-generation lung cancer treatment. Continued research focusing on large-scale manufacturing, long-term safety evaluation, regulatory considerations, and clinical translation will be essential to realize the full potential of these advanced nanomedicine platforms in precision oncology and personalized cancer therapy..
REFERENCES

Mehetre Rutuja, Dr. Sachin Aglave, Dr. Vijay Jadhav, Dr. Nitin Jain, Dr. Usha Jain, Oroxylin A-Loaded Zwitterionic Mesoporous Silica Nanoparticles: A Promising Platform for Targeted Lung Cancer Therapy, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 507-535. https://doi.org/10.5281/zenodo.21154768
 10.5281/zenodo.21154768
10.5281/zenodo.21154768