
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Guru Nanak College of Pharmaceutical Sciences, Dehradun
Modern drug development is an extraordinarily complex, costly, and protracted endeavor, requiring 10–15 years and $1–2.6 billion to bring a single new molecular entity to market, with the majority of candidates failing due to insufficient efficacy or unacceptable toxicity. This high-risk landscape has evolved dramatically from ancient folklore remedies and serendipitous discovery (e.g., aspirin, penicillin) to the mid-20th century rise of empirical in vivo screening, and then to the molecular biology revolution of the 1980s–1990s, which ushered in target based discovery exemplified by imatinib and HIV protease inhibitors. Today, the field has entered an integrated, multi omics, data driven era, leveraging genomics, proteomics, and CRISPR-based gene editing to identify and de risk targets before a single compound is synthesized. Target identification begins with unbiased genetic or transcriptomic data—such as GWAS revealing PCSK9 as a cardiovascular target or cancer genomics uncovering driver mutations like BRAF V600E—to pinpoint a causal molecular entity. Yet even a biologically relevant target may fail if it is not “druggable.” Classical druggable families (GPCRs, ion channels, kinases, nuclear receptors) constitute only 10–15% of the human proteome; the remainder, including Ras, transcription factors, and protein protein interaction interfaces, have long been deemed “undruggable,” though advances in covalent inhibitors, PROTACs, and RNA-targeting modalities are progressively breaching these barriers. Rigorous target validation—using orthogonal genetic (CRISPR knockout/knockin) and chemical (tool compound) approaches—is essential to confirm therapeutic relevance before advancing to lead discovery. Key enabling technologies include whole genome sequencing, single cell transcriptomics, in vivo CRISPR screens, and organ on chip models. Together, these foundational steps of target identification, druggability assessment, and multi level validation form the indispensable bedrock upon which successful drug development is built, tempering enthusiasm for new targets with the sobering realities of attrition and cost.
The scope and cost of modern drug development are staggering, representing one of the most complex, highly regulated, and financially intense endeavors in human enterprise[1]. To bring a single new molecular entity to the pharmacy shelf, a pharmaceutical company must navigate a decade‐long odyssey that typically consumes between 1billionand 2.6 billion in research and development expenditures, with some estimates including the cost of failed candidates pushing the figure above 2 million, and a pivotal Phase 3 program can easily exceed $100 million. In recent years, the cost trajectory has been further inflated by the shift toward precision medicines, gene therapies, and biologics, which require specialized manufacturing and often target smaller patient subpopulations, thereby reducing potential revenue while not proportionally lowering development costs. It is against this daunting economic and scientific backdrop that the historical evolution of drug discovery must be appreciated. Before the modern era, pharmacopoeias were essentially compilations of folklore, serendipity, and trial‐and‐error observation. Ancient remedies derived from willow bark (the origin of salicylic acid, later acetylated to become aspirin), foxglove (digitalis), and cinchona bark (quinine) were used for centuries without any knowledge of their molecular targets. The nineteenth century brought the first true paradigm shift: the isolation and purification of active principles from natural sources, such as morphine from opium (1804) and penicillin’s chance observation by Fleming in 1928, followed by the Florey–Chain team’s mass production[2]. Yet drug discovery remained largely empirical until the mid‐twentieth century, when the advent of organic chemistry enabled systematic modification of lead compounds. The 1950s–1970s saw the rise of high‐throughput in vivo screening, where thousands of chemicals were tested in animal models of disease—a process that was effective but slow and ethically demanding. A transformative leap occurred with the molecular biology revolution of the 1980s and 1990s. The ability to clone, express, and purify human proteins enabled target‐based drug discovery: instead of screening compounds in whole animals, researchers could design assays against a specific enzyme or receptor implicated in disease. This rational, reductionist approach was hailed as a new dawn, leading to blockbusters like the protease inhibitors for HIV and the tyrosine kinase inhibitor imatinib (Gleevec) for chronic myeloid leukemia. However, the target‐based paradigm also revealed its own limitations, including the challenge of validating that hitting a target in a test tube truly translates to therapeutic benefit in a patient. The twenty‐first century has ushered in an era of integrated, multi‐omics, and data‐driven discovery, where genomics, proteomics, and gene‐editing technologies like CRISPR are used not only to find targets but also to de‐risk them before a single compound is synthesized[3]. Understanding this historical evolution is crucial because the methods used to identify and validate a drug target—the biological origin of disease—are the most critical determinants of subsequent success. Target identification begins with a deep investigation of disease pathophysiology. For a given condition—say, a form of non‑small cell lung cancer driven by an epidermal growth factor receptor (EGFR) mutation—the goal is to pinpoint a specific molecular entity (protein, nucleic acid, or even a metabolic pathway) whose modulation is likely to produce a therapeutic effect[4]. Historically, targets were often identified through phenotypic observations: a compound that lowered blood pressure in an animal model led researchers to work backward to discover its mechanism (e.g., calcium channel blockade). Today, the starting point is frequently unbiased genetic or transcriptomic data. Genome‑wide association studies (GWAS) can link single‑nucleotide polymorphisms to disease risk; for example, variants in the gene PCSK9 were found to correlate with low cholesterol levels and protection against coronary artery disease, immediately suggesting PCSK9 as a therapeutic target. Similarly, cancer genomics has revealed driver mutations such as BRAF V600E in melanoma and BCR‑ABL in leukemia. Beyond genetics, proteomics and metabolomics can identify proteins or metabolites that are consistently altered in diseased versus healthy tissue. The ideal target is one that is causally linked to the disease, is amenable to modulation by a drug‐like molecule, and, when modulated, produces a sufficient margin between desired therapeutic effects and on‑target toxicity. However, identifying a candidate is only the first step; the vast majority of targets fail not because they are irrelevant but because they are not “druggable”—a concept that lies at the heart of druggability assessment and the challenge of “undruggable” targets. Druggability refers to the likelihood that a target protein can be bound by a small molecule or biologic with high affinity and selectivity under physiological conditions[5]. For classical drug targets like G‑protein coupled receptors (GPCRs), ion channels, kinases, and nuclear hormone receptors, the druggable genome has been well charted: about 10–15% of the human proteome falls into these families. But the remaining 85%—including transcription factors, scaffolding proteins, protein–protein interaction interfaces, and many enzymes without deep hydrophobic pockets—have historically been deemed undruggable. The classic example is the Ras family of small GTPases, mutated in nearly 30% of human cancers; for decades Ras was considered undruggable because it lacks a well‑defined binding pocket and binds to its natural ligand GTP with picomolar affinity, making competitive inhibition nearly impossible[6]. Similarly, MYC, p53, and other transcription factors were dismissed as “undruggable” because they are intrinsically disordered and lack active sites. Fortunately, recent technological advances have begun to breach this fortress. Fragment‑based drug discovery has identified shallow, cryptic pockets on Ras proteins that can be occupied, leading to the first approved KRAS G12C inhibitors (sotorasib, adagrasib)[7]. Covalent inhibitors, which form permanent bonds with reactive cysteine residues, have enabled targeting of previously intractable enzymes. Beyond small molecules, new modalities have expanded the druggable universe: proteolysis‐targeting chimeras (PROTACs) harness the cell’s ubiquitin‑proteasome system to degrade any protein of interest, irrespective of its druggability; RNA‑targeting small molecules and antisense oligonucleotides can modulate non‑coding RNAs or splicing; and cell and gene therapies directly address genetic deficiencies. Thus, while the concept of “undruggability” remains a real barrier, it is increasingly a moving target[9]. Even with a potentially druggable candidate in hand, the critical next step—target validation—must confirm that modulating that target is therapeutically relevant. Validation is the process of accumulating multiple, orthogonal lines of evidence that altering the target’s function (inhibiting an enzyme, blocking a receptor, reducing protein levels) leads to a desired phenotypic change in disease models without unacceptable off‑effects. The gold standard for genetic validation is loss‑of‑function (LOF) and gain‑of‑function (GOF) studies in model organisms or human cells. For decades, this meant generating knockout mice or using RNA interference (RNAi)[10]. However, RNAi often suffers from off‑target effects and incomplete knockdown, leading to false positives. The CRISPR‑Cas9 revolution has transformed target validation by enabling precise, efficient, and scalable gene editing. Using CRISPR, researchers can knock out a single gene in human cell lines, patient‑derived organoids, or even entire zebrafish or mice, then assess the impact on disease‑relevant phenotypes. Moreover, CRISPR interference (CRISPRi) and activation (CRISPRa) allow tunable, reversible modulation without cutting DNA, mimicking the effect of a drug more closely. Complementary chemical validation is equally important: if a tool compound—a selective pharmacological modulator of the target—can recapitulate the genetic phenotype, confidence in the target increases dramatically[11]. Ideally, genetic and chemical validation converge: for instance, the PCSK9 story was validated both by human genetics (individuals with LOF mutations have low LDL and no increased disease risk) and by monoclonal antibodies (evolocumab, alirocumab) that lower LDL in patients. A validated target is one where the causal chain from target modulation to disease modification has been established, the safety margin is estimated, and the biological context is understood (e.g., compensatory pathways, tissue‑specific expression). However, over‑reliance on reductionist models can be misleading; a target validated in a cell line may fail in an intact organism due to pharmacokinetic issues or unrecognized homeostatic responses[12]. Therefore, modern validation increasingly incorporates human genetics (Mendelian randomization, colocalization analyses), patient‑derived induced pluripotent stem cell (iPSC) models, and organ‑on‑chip systems that recapitulate multi‑cellular architecture. Only after rigorous target validation does the project transition into hit identification—but even the most sophisticated screening campaign cannot rescue a poorly validated target[12]. The key technologies that enable both target identification and validation have exploded in sophistication over the past two decades. Genomics, first and foremost, has moved from microarrays to whole‑exome and whole‑genome sequencing at ever‑diminishing costs. The availability of large‑scale biobanks (UK Biobank, All of Us) combined with electronic health records allows phenome‑wide association studies (PheWAS) that can reveal unexpected on‑target side effects or repurposing opportunities. Proteomics, once limited by sensitivity, now uses mass spectrometry and affinity‑based methods to quantify thousands of proteins across tissues, while spatial proteomics maps protein localization within tissue architecture. The integration of genomic and proteomic data—proteogenomics—can identify neoantigens in cancer or confirm that genetic variants lead to changes in protein abundance or function. But perhaps no technology has transformed target validation as profoundly as CRISPR‑Cas9. Beyond simple knockouts, CRISPR screens have become a routine tool for functional genomics. Pooled CRISPR libraries, containing tens of thousands of single‑guide RNAs targeting every gene in the genome, can be transduced into cells, and after positive or negative selection (e.g., survival after drug treatment), deep sequencing reveals which gene knockouts confer resistance or sensitivity. Such screens are used not only to identify essential genes in cancer but also to discover resistance mechanisms to existing drugs, thereby suggesting combination targets[13]. Base editing and prime editing now allow the introduction of specific disease‑associated point mutations without double‑strand breaks, enabling more precise validation of non‑synonymous variants. Moreover, in vivo CRISPR screens, performed directly in animal models, can validate targets in a more physiologically relevant context, for instance, by delivering CRISPR libraries to the liver or brain via viral vectors. Complementing CRISPR, advances in single‑cell RNA sequencing (scRNA‑seq) have revealed that what once looked like a homogeneous disease population is often a mosaic of cellular states; a target may be relevant only in a rare subpopulation of cells, explaining why bulk analyses failed. Epigenomic technologies (ATAC‑seq, ChIP‑seq) illuminate the regulatory landscape, identifying non‑coding regions that might be targeted by small molecules or antisense oligonucleotides[14]. Together, these technologies form a virtuous cycle: genomics suggests targets, CRISPR validates causality, proteomics confirms protein expression, and single‑cell methods contextualize the target within heterogeneous tissues. Consequently, the modern drug hunter no longer begins with a chemical library but with a deep biological question, armed with a toolkit that would have seemed like science fiction just a generation ago. Yet, for all this power, the sobering reality remains that even the most beautifully identified and validated target can still fail in the clinic if the drug candidate does not achieve sufficient exposure at the site of action, if animal models poorly recapitulate human disease, or if the target’s biology is more complex than anticipated. Thus, the journey from biological origin to approved medicine is a marathon that demands not only technological prowess but also rigorous integration of target identification, druggability assessment, and multi‑level validation—steps that together form the indispensable foundation upon which all subsequent stages of drug development are built[15][16].
THE DISCOVERY PHASE: FROM HITS TO LEADS
Hit Identification: High‑Throughput and Virtual Screening – The journey from a validated biological target to a drug candidate begins with hit identification, the process of discovering chemical matter that shows reproducible activity against the target. Two complementary strategies dominate this landscape: high‑throughput screening (HTS) and virtual screening (VS). High‑throughput screening is the workhorse of the pharmaceutical industry, enabling the testing of hundreds of thousands to millions of compounds against a purified target protein or a cell‑based phenotype in an automated, miniaturized format. Modern HTS facilities can evaluate over 100,000 compounds per day using robotic liquid handlers, sensitive detectors (fluorescence, luminescence, or label‑free technologies), and sophisticated data management systems[17]. The compound libraries used in HTS have evolved from simple collections of drug‑like molecules to include diverse chemotypes, fragments, natural product derivatives, and even DNA‑encoded libraries (DELs) that can screen billions of compounds simultaneously through affinity selection and sequencing. However, HTS is resource‑intensive and requires large quantities of target protein, a robust and reproducible assay, and significant infrastructure. In contrast, virtual screening offers a computational alternative that can rapidly sift through vast chemical databases—sometimes exceeding 10 billion compounds—without physically synthesizing or purchasing them. Virtual screening falls into two broad categories[18]: ligand‑based and structure‑based. Ligand‑based virtual screening uses known active molecules (a pharmacophore or quantitative structure‑activity relationship model) to search for structurally similar compounds in databases; this approach is useful when the three‑dimensional structure of the target is unknown. Structure‑based virtual screening, often called molecular docking, requires a high‑resolution crystal structure or a reliable homology model of the target. Each compound from a database is computationally placed into the target’s binding site, and scoring functions estimate binding affinity, complementarity, and desolvation penalties[19][20].
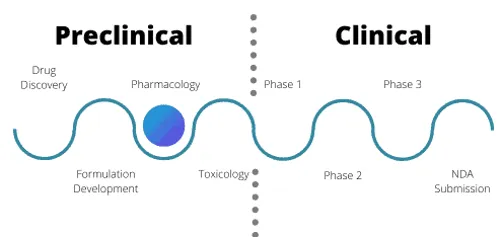
Fig: 1 Drug discovery from phase 1 to phase 4
While docking is fast—millions of compounds can be processed in days—its accuracy is limited by protein flexibility, water molecules, and scoring function approximations. Thus, successful hit identification increasingly employs a hybrid approach: virtual screening pre‑filters a large database to a manageable set of a few thousand compounds, which are then tested experimentally using HTS or a focused biochemical assay. Hits are defined by stringent criteria: typically, greater than 50% inhibition at a single concentration (e.g., 10 µM) followed by confirmation in dose‑response curves to derive IC₅₀ values. At this stage, hits are rarely potent (often low‑micromolar) and may show poor physicochemical properties, but they serve as the starting points for the next phase. Hit‑to‑Lead Transition and Chemical Space Exploration – Once a set of confirmed hits is in hand, the project enters the hit‑to‑lead (H2L) phase, whose goal is to identify a smaller number of chemically distinct series—usually 2 to 5—that have sufficient potency (IC₅₀ < 1 µM, often < 100 nM), preliminary selectivity over related targets, and acceptable in vitro ADME properties (microsomal stability, plasma protein binding, CYP inhibition, and early permeability or solubility screens)[21]. The transition from hit to lead is not merely a filtering exercise; it is an exploratory voyage through chemical space. Chemical space is the theoretical universe of all possible small molecules, estimated to contain up to 10⁶⁰ compounds—an impossibly vast number. In practice, medicinal chemists navigate a tiny fraction of that space by synthesizing analogues around a promising hit scaffold[22]. Hit‑to‑lead activities involve rapid analogue synthesis (e.g., replacing substituents, ring scissions or fusions, bioisosteric replacements) guided by preliminary SAR. The aim is to achieve a “lead” that is not yet optimized but has demonstrated a clear structure‑activity relationship, evidence of cellular activity (if the target is intracellular), an initial pharmacokinetic profile in rodents (often just single‑time‑point exposure), and confirmatory binding or functional data. Crucially, the H2L phase also evaluates patentability and developability[23]. A lead series must have a novel scaffold or a new mode of interaction to allow freedom‑to‑operate; otherwise, the project may be abandoned. Chemical space exploration during this stage benefits from fragment‑based approaches, where very small, low‑affinity fragments (MW < 300) are identified by biophysical methods (NMR, surface plasmon resonance, X‑ray crystallography) and then elaborated or linked into larger, more potent leads[24]. Additionally, diversity‑oriented synthesis and DNA‑encoded chemistry have expanded the accessible chemical space, enabling the discovery of novel chemotypes that would not appear in traditional HTS collections. At the end of hit‑to‑lead, one or two lead series are selected for intensive optimization. Lead Optimization: Enhancing Potency, Selectivity, and ADME Properties – Lead optimization (LO) is the most resource‑intensive medicinal chemistry effort, where the goal is to convert a lead compound—typically with low‑micromolar to high‑nanomolar potency, some selectivity issues, and suboptimal drug metabolism and pharmacokinetic (DMPK) properties—into a preclinical candidate suitable for formulation, toxicology, and eventual clinical trials. The process is iterative: design, synthesis, biological testing, and DMPK profiling cycles are repeated dozens or hundreds of times, with each cycle aiming to improve multiple parameters simultaneously while avoiding the introduction of new liabilities. Potency enhancement is often achieved by deepening understanding of the target’s binding site: X‑ray co‑crystal structures or cryo‑EM models guide the introduction of functional groups that form hydrogen bonds, hydrophobic contacts, or π‑stacking interactions. However, blindly increasing potency can lead to increased molecular weight and lipophilicity, which often worsens solubility and promotes off‑target toxicity[25]. Selectivity over closely related enzymes or receptors (e.g., selectivity among kinases, GPCRs, or cytochrome P450s) is equally critical. A kinase inhibitor that hits dozens of off‑target kinases may cause unacceptable side effects; therefore, selectivity is engineered by exploiting subtle differences in the binding pocket’s shape, flexibility, or amino acid composition. Beyond potency and selectivity, ADME properties become the dominant focus during lead optimization. Absorption depends on solubility and permeability, which often trade off against each other. Distribution is influenced by plasma protein binding, tissue accumulation, and the compound’s ability to cross the blood‑brain barrier if a central nervous system (CNS) drug is desired—or to avoid crossing it for peripheral targets[26]. Metabolism, primarily by cytochrome P450 enzymes, dictates half‑life and clearance; medicinal chemists strategically block metabolic soft spots (e.g., by adding fluorine atoms to prevent oxidation of a benzylic carbon) or reduce CYP inhibition to avoid drug‑drug interactions. Excretion and potential for drug‑induced liver injury are assessed via reactive metabolite trapping studies and in vitro hepatocyte assays[27]. The ultimate output of lead optimization is a development candidate that demonstrates acceptable efficacy in animal models of disease, a favorable safety margin in toxicology studies, and a consistent pharmacokinetic profile enabling once‑ or twice‑daily dosing. Structure‑Activity Relationships (SAR) and Drug‑likeness (Rule of Five) – The intellectual engine driving both hit‑to‑lead and lead optimization is the systematic exploration of structure‑activity relationships (SAR). SAR is the empirical correlation between the chemical structure of a molecule and its biological activity, often expressed as changes in IC₅₀, EC₅₀, or other functional endpoints as specific substituents are varied. In a typical SAR table, a series of analogues is arranged with columns for R‑group modifications and rows for biological and physicochemical data; medicinal chemists look for trends—for example, adding a small hydrophobic group at a certain position improves potency, but a polar group destroys it[28]. Modern SAR is increasingly multi‑parametric, tracking not just potency but also metabolic stability, permeability, solubility, and off‑target effects (e.g., hERG channel inhibition, which can cause cardiac arrhythmia). As SAR matures, quantitative SAR (QSAR) models use computational algorithms to predict activity and properties from molecular descriptors. While SAR defines what structural features are required for target engagement, the concept of drug‑likeness—encapsulated by Lipinski’s “Rule of Five” (Ro5)—sets boundaries for oral bioavailability. The Rule of Five states that poor absorption or permeation is more likely when a molecule violates two or more of the following criteria: molecular weight > 500, calculated log P (clogP) > 5, hydrogen bond donors (HBD, sum of –OH and –NH groups) > 5, and hydrogen bond acceptors (HBA, sum of N and O atoms) > 10. The rule emerged from analysis of over 2,000 drug candidates that had entered Phase 2 clinical trials; it is a heuristic, not a law, but it has proven remarkably durable. More than 90% of orally administered drugs that have reached the market conform to the Ro5. However, modern drug discovery has generated many exceptions, particularly for natural products (e.g., ivermectin, MW ~875) and for drugs that are substrates of active transporters (e.g., statins) or that are injected (biologics, peptides). Thus, the Rule of Five is best used as a filter during hit‑to‑lead and lead optimization to avoid compounds with predictably poor permeability or solubility, but with the understanding that violations can be compensated by high potency, transporter uptake, or formulation strategies. In summary, the seamless integration of high‑throughput and virtual screening, careful navigation of chemical space during hit‑to‑lead transition, multiparameter optimization of potency, selectivity and ADME properties, and the disciplined application of SAR and drug‑likeness rules collectively transform a hit into a robust preclinical candidate, ready to face the rigors of in vivo toxicology and, ultimately, clinical trials.
4. PRECLINICAL DEVELOPMENT
Once a lead compound has been optimized for potency, selectivity, and drug‑likeness, it enters the preclinical development phase, where the central question shifts from “Does it hit the target?” to “Does it produce a therapeutic effect in a living system, and is it safe enough to test in humans?” This phase is anchored by four interlocking pillars: pharmacodynamics (PD), pharmacokinetics (PK) with its ADME components, toxicology and safety pharmacology, and formulation development.
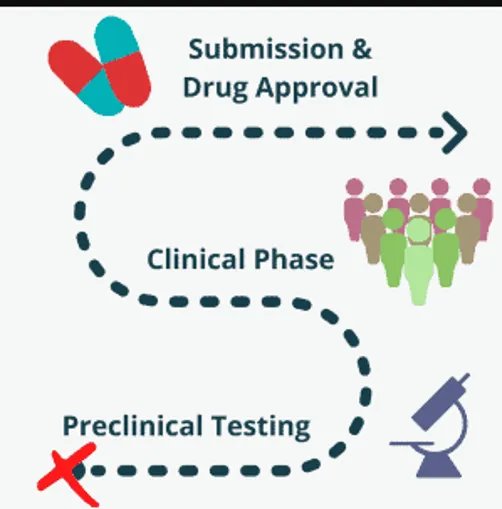
Fig: 2 Preclinical developments from submission to testing
The integration of these pillars ultimately culminates in the Investigational New Drug (IND) application, the regulatory gateway to clinical trials[29][30]. Pharmacodynamics, in both its in vitro and in vivo manifestations, describes what the drug does to the body—that is, the relationship between drug concentration at the site of action and the resulting pharmacological effect. In vitro PD studies begin with cell‑based assays using the target cell type (e.g., cancer cell lines, primary hepatocytes, or neurons) to measure functional responses such as enzyme inhibition, receptor activation, second‑messenger modulation, or gene expression changes. Concentration‑response curves yield key parameters like EC₅₀ (half‑maximal effective concentration), IC₅₀ (half‑maximal inhibitory concentration), and Emax (maximal effect). Beyond simple potency, in vitro PD explores efficacy in disease‑relevant phenotypes: for an anti‑cancer compound, this means apoptosis induction or proliferation arrest; for an anti‑infective, minimum inhibitory concentration (MIC) against bacterial or fungal pathogens[31]. However, in vitro PD often occurs in artificial, reductionist environments—cells in plastic wells without immune components, blood flow, or three‑dimensional architecture. Therefore, in vivo PD studies in animal models are indispensable. Rodent models of disease (xenograft tumors, induced arthritis, transgenic Alzheimer’s mice) are used to measure pharmacodynamic endpoints after dosing the candidate compound. Typical in vivo PD readouts include tumor volume reduction, blood pressure lowering, blood glucose normalization, or improvement in behavioral scores. Crucially, in vivo PD establishes a temporal and dose‑dependent relationship between drug administration and effect, often revealing non‑linearities such as ceiling effects or rebound phenomena. The integration of in vivo PD with pharmacokinetics—a discipline that studies what the body does to the drug—is so fundamental that it has given rise to pharmacokinetic/pharmacodynamic (PK/PD) modeling, which links drug exposure to effect magnitude and duration. Pharmacokinetics encompasses the four ADME processes: absorption, distribution, metabolism, and excretion. Preclinical PK profiling begins with in vitro ADME assays that are high‑throughput and informative. Absorption is initially assessed using parallel artificial membrane permeability assays (PAMPA) or Caco‑2 cell monolayers (which model intestinal absorption and also measure active efflux by P‑glycoprotein). Distribution parameters are estimated from plasma protein binding (equilibrium dialysis), blood‑to‑plasma ratio, and early tissue distribution studies in rodents. Metabolism profiling includes liver microsome or hepatocyte stability experiments to determine intrinsic clearance, metabolite identification using high‑resolution mass spectrometry, and cytochrome P450 (CYP) inhibition and induction assays to predict drug‑drug interactions. Excretion pathways are inferred from mass balance studies and transporter interaction assays (e.g., BCRP, OATP1B1). These in vitro data are then used to design in vivo PK studies, typically in rodents and a non‑rodent species (often dogs or minipigs). Animals receive the compound by intravenous (IV) and oral (PO) routes; serial blood samples are collected, and plasma concentrations are analyzed by LC‑MS/MS. Non‑compartmental analysis yields key PK parameters: half‑life (t₁/₂), clearance (CL), volume of distribution (Vd), area under the curve (AUC), peak concentration (Cmax), time to peak (Tmax), and oral bioavailability (F)[32][33]. A compound with poor bioavailability (<20%) may still be viable for parenteral administration, but for an oral drug, bioavailability of at least 50% is often targeted. Importantly, PK/PD integration involves measuring drug concentrations in the effect compartment (e.g., tumor tissue, brain, synovial fluid) rather than just plasma; sometimes plasma exposure correlates poorly with target engagement. Allometric scaling of PK data from animals to humans is then performed, though its accuracy is limited and species differences (e.g., in metabolism) can mislead[34]. While PD and PK ask whether the drug works, toxicology and safety pharmacology ask whether it can be given safely. Regulatory guidelines (ICH M3, S7A, S9) mandate a battery of toxicology studies in at least two mammalian species (one rodent, one non‑rodent) before a candidate can enter human trials[35]. The core of toxicology is the repeated‑dose toxicity study, where animals are treated with the drug at multiple dose levels (including a vehicle control and a high‑dose that produces overt toxicity) for durations that match the intended clinical trial length—typically 2 weeks to 3 months for Phase 1. Endpoints include mortality, clinical observations, body weight, food consumption, clinical pathology (hematology, serum chemistry, coagulation), urinalysis, organ weights, and gross and microscopic histopathology of every major organ. The no‑observed‑adverse‑effect level (NOAEL) and lowest‑observed‑adverse‑effect level (LOAEL) are determined[36][37]. Genotoxicity is assessed using the Ames test (bacterial reverse mutation), in vitro micronucleus or chromosome aberration assays, and an in vivo micronucleus test. If the drug is intended for chronic use, a carcinogenicity study (2 years in rodents) is required later, but is not needed for initial IND. Safety pharmacology studies evaluate the potential for undesirable effects on vital organ systems, independent of the intended pharmacodynamics. The core battery includes cardiovascular safety (hERG channel inhibition in vitro, followed by telemetry in conscious animals to measure blood pressure, heart rate, and ECG intervals—especially QTc prolongation), central nervous system safety (functional observation battery assessing locomotor activity, behavior, and seizure potential), and respiratory safety (plethysmography to measure respiratory rate and tidal volume). Additional safety pharmacology studies may be needed for renal, gastrointestinal, or other systems if there are specific concerns. All toxicology and safety pharmacology studies must be conducted under Good Laboratory Practice (GLP) regulations to ensure data integrity and reproducibility. The final pillar of preclinical development is formulation development and route of administration. The lead compound is rarely administered as pure powder; it must be formulated into a stable, bioavailable, and reproducible dosage form[38]. For early toxicology studies, a simple solution, suspension, or excipient blend is often sufficient. However, for the IND‑enabling GLP toxicology study and the planned Phase 1 clinical trial, the formulation should be representative of the intended clinical product. Formulation scientists must consider solubility, stability (chemical and physical), pH, osmolality, sterility (for parenterals), and compatibility with administration devices. For oral formulations, they may develop capsules, tablets, or amorphous solid dispersions to enhance solubility[39]. For intravenous administration, they produce sterile solutions or lyophilized powders that are reconstituted before infusion. The route of administration is dictated by the target indication and PK properties. Oral is preferred for chronic, out‑patient use, but compounds with very low bioavailability or first‑pass metabolism may require intravenous, subcutaneous, or intramuscular injection[40]. Other routes—topical, inhaled, intrathecal (for CNS), or ocular—are used for local delivery. The formulation must be scaled up from bench (grams) to pilot scale (kilograms) to support the toxicology studies and then to manufacturing scale for clinical trials. All of these preclinical data—PD, PK/ADME, toxicology, safety pharmacology, and formulation—are assembled into the Investigational New Drug (IND) application, the formal request to the FDA (or equivalent global agency) to begin human clinical trials. The IND is a massive document, typically thousands of pages, organized into seven sections: introductory statement and investigational plan, investigator’s brochure, clinical protocol, chemistry manufacturing and control (CMC) data, pharmacology/toxicology data (including all GLP study reports), previous human experience (if any), and additional information[41]. The core pharmacology/toxicology section must demonstrate that the candidate has sufficient activity in relevant animal models, a NOAEL that supports a safe starting dose in humans (usually calculated as 1/10th to 1/50th of the NOAEL in the most sensitive species), and no unacceptable risks for the proposed clinical study. Once the IND is submitted, the FDA has 30 days to review it; if no “clinical hold” is issued, the sponsor may proceed with Phase 1 trials[42][43]. The IND is not a single approval but a living application that is updated as new data emerge. In summary, the preclinical phase transforms a chemical lead into a regulated, safe, and well‑characterized drug candidate, with PD proving it works, PK/ADME defining its exposure, toxicology and safety pharmacology establishing its margins of safety, formulation providing a practical dosage form, and the IND application integrating everything into a legal and scientific dossier that unlocks the door to human testing[44][45].
CONCLUSION
The drug development pipeline is a meticulously staged journey, beginning with target discovery and validation, moving through hit identification, lead optimization, and preclinical safety assessment, then advancing into three phases of clinical trials, regulatory review, and finally post-marketing surveillance. Each gate is designed to filter out unsafe or ineffective candidates before they reach patients, acknowledging that fewer than 10% of initial hits ultimately receive approval. This linear yet iterative process balances the urgent need for new therapies against the non‑negotiable imperatives of safety and efficacy. In final remarks, the triad of safety, efficacy, and innovation remains the cornerstone of pharmaceutical progress. Safety demands rigorous toxicology and pharmacovigilance, while efficacy requires robust proof from well‑controlled trials. Innovation—whether through AI‑driven discovery, gene editing, or novel modalities like PROTACs—must accelerate development without compromising the other two pillars. The rising costs and attrition rates underscore that shortcuts are perilous, yet the human toll of untreated diseases equally compels urgency. Ultimately, a successful drug is not simply a potent molecule but a thoroughly vetted therapeutic that demonstrably improves patient outcomes, embodying the harmonious integration of scientific ingenuity, regulatory rigor, and ethical responsibility.
REFERENCES

Soumyadeep Dutta, Mohit Gupta, Stage of Drug Discovery and Drug Development Process, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 429-442. https://doi.org/10.5281/zenodo.20501626
 10.5281/zenodo.20501626
10.5281/zenodo.20501626