
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Chemistry, College of Pharmaceutical Sciences,Government Medical college, Thiruvananthapuram.
Thiazoles are unique five-membered aromatic heterocycles with electron-donating sulfur and electron-withdrawing nitrogen atoms and are a highly privileged scaffold in modern medicinal chemistry. These compounds have a well-established role in therapeutics (e.g. penicillin, ritonavir and bleomycin) and provide an exceptionally versatile foundation for rational drug design. This review is about the broad pharmacological potential of newly synthesized thiazole derivatives and hybrid molecules against a wide range of disease targets. Researchers have successfully obtained advanced drug candidates with significantly enhanced efficacy, target specificity and metabolic stability by actively utilizing molecular hybridization strategies, such as the fusion of thiazole with structurally diverse moieties, such as pyrazole, quinazoline and adamantane. The review systematically emphasizes the remarkable anti-cancer activity of these derivatives, including their ability to induce cell cycle arrest and apoptosis, and selectively inhibit important kinases such as VEGFR-2, EGFR and CDKs. Furthermore, thiazole hybrids have exhibited potent antimicrobial and antiviral activities, where they can effectively combat multi-drug resistant bacterial strains (e.g. MRSA) and inhibit essential replication enzymes of pathogens such as the Foot-and-Mouth Disease Virus and coronaviruses
Thiazoles are important nitrogen-containing aromatic heterocyclic compounds possessing various biological activities and their usefulness as medicines are well recognized. These compounds have been examined for their significant antimicrobial activity against a variety of clinically relevant bacterial and fungal strains. These studies have concluded that thiazole ring is a good pharmacophore for the design of bioactive molecules. For instance, thiazole containing molecules such as sulfathiazole are used as an antimicrobial drug, ritonavir as antiretroviral drug, abafungin as antifungal drug and bleomycine and thiazofurin as antineoplastic drug. Thiazole is also present in penicillin which is the first one of the broad-spectrum antibiotics1.
Thiazoles represent one of the most extensively studied classes of five-membered aromatic heterocycles in medicinal chemistry. Over the past three decades, research has consistently demonstrated that the thiazole scaffold possesses a broad spectrum of biological activities, including antioxidant, antibacterial, antiviral, diuretic, antitumor, and anticonvulsant properties. Structurally, the thiazole ring is unique due to the presence of both an electron-donating Sulfur atom and an electron-accepting nitrogen atom. Its aromaticity arises from the delocalization of a lone pair of electrons from the Sulfur atom into the ring's pi system, which completes the electron sextet required to satisfy Hückel's rule2.
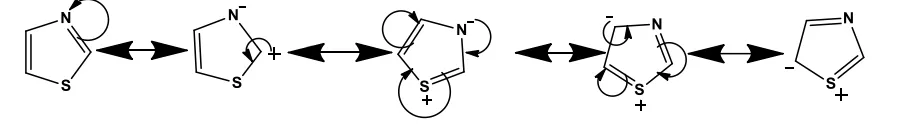
Figure 1: Resonating structure of thiazole2 .
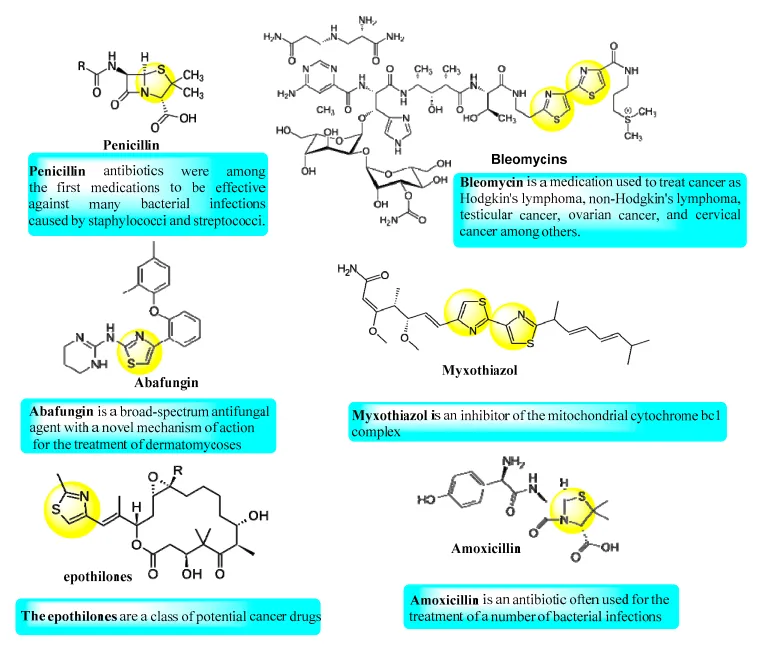
Figure 2 : Natural and available marketing drugs containing thiazole rings3.
Thiazole is a key constituent of azole heterocycles, which also include pyrazoles, imidazoles, isoxazoles, and oxazoles. It is an aromatic heterocyclic organic compound featuring a five-membered ring that contains both a nitrogen atom and a sulfur atom, with the molecular formula C3H3NS. Thiazole is found in various specific compounds, often fused with a benzene ring, known as benzothiazoles. Structurally, thiazole is related to imidazole, where sulfur is replaced by nitrogen, and oxazole, where sulfur is replaced by oxygen. The nitrogen and sulfur atoms in thiazole, an important aromatic five-membered heterocycle, are responsible for its unique biological properties. According to previous studies, thiazole-containing compounds exhibit a wide range of biological activities, including:
THIAZOLE DERIVATIVES AS ANTICANCER AGENTS
Cancer is a disease characterized by abnormal uncontrolled cell cycle progression and accelerated proliferation of normal cells Cancer has been identified as the world’s second greatest cause of mortality with an year after year, the rising number of new instances of cancer, preceded only by heart illness. Cancer-related death rates are increasing an unparalleled speed in many emerging countries. Cancer is not just a health problem, but also a social and economic one.The estimated annual economic cost of cancer in 2010 was roughly 1.16 trillion. The total cost of cancer in the United States is anticipated to reach 173 billion in 20204.
Recently, Hussein et al. (2024) explores the development of novel pharmaceutically active compounds. The researchers aimed to synthesize a new series of integrated thiazole-pyrazole derivatives and evaluate their potential as both antioxidant and anticancer agents. The new derivatives were synthesized through an efficient reaction between hydrazonoyl chloride derivatives and a carbothioamide derivative. Their structural identities were confirmed using spectroscopic techniques (IR, NMR, Mass spectrometry) and elemental analysis. Density Functional Theory (DFT) was utilized to optimize the structures and minimize their energy. The compounds' antioxidant capabilities were tested using DPPH and ABTS radical scavenging assays. Their cytotoxicity was evaluated against HepG2 (hepatocellular carcinoma) cell lines using the MTT assay. The compounds were computationally docked against the tyrosine kinase receptor (PDB ID: 2HCK) using MOE software to understand their binding interactions at a molecular level. All tested compounds exhibited strong anti-tumor activity against the HepG2 cell lines. Derivatives 1a and 1b were the most potent, demonstrating the lowest IC50 values (1.324 µg/mL and 1.398 µg/mL, respectively). The study found that incorporating phenyl, p-tolyl, and p-nitro groups into the molecular structure enhanced the compounds' cytotoxic efficacy compared to p-chloro analogues. The synthesized compounds showed strong ABTS scavenging activity that was comparable to the standard antioxidant, gallic acid, particularly at higher concentrations. However, their DPPH scavenging activity was only moderate. The derivatives displayed significantly higher binding energies (ranging from -7.56 to -8.89 kcal/mol) when docked against tyrosine kinase compared to quercetin, the original reference ligand (-5.92 kcal/mol)5.


Al-Salmi et al. (2023) reported synthesis, characterization and biological evaluation of a novel series of thiazole derivatives targeting cancer cell proliferation. The study shows the potentiality of these compounds as effective chemotherapeutic agents especially in breast cancer. A new series of 2-[2-[4-hydroxy-3-substituted benzylidene hydrazinyl]-thiazole-4[5H]-ones and an acetoxy derivative have been synthesized. The synthesis was carried out by using thiosemicarbazones (2a-c). The molecular structures of all newly synthesized derivatives were confirmed by elemental investigations and spectroscopic methods such as IR and NMR. The biological activity of the synthesized compounds was evaluated based on their ability to suppress the growth of MCF-7 (breast) and HepG2 (liver) cancer cell lines. Compound 2 (featuring an R=N=N-Ph substitution) demonstrated the highest cytotoxic efficacy, outperforming the standard drug Staurosporine in both tested cell lines. Because of its superior cytotoxic profile, compound 2 was subjected to further mechanistic evaluation in MCF-7 cells: VEGFR-2 Inhibition: Compound 2 effectively blocked the vesicular endothelial growth factor receptor-2 (VEGFR-2) with an IC50 of 0.15 µM, showing moderate effectiveness compared to the standard Sorafenib IC50 = 0.059 µM. Cell Cycle Arrest: Flow cytometry analysis revealed that compound 2 induced cell cycle arrest at the G1/S phase. DNA Accumulation: Treatment increased the accumulation of MCF-7 cells in the pre-G1 phase to 37.36%, compared to just 2.02% in the untreated control. Apoptosis Induction: Compound 2 significantly triggered apoptosis, increasing early apoptosis to 22.39% and late apoptosis to 9.51% (up from 0.51% and 0.29% in untreated cells, respectively). The compound also increased cell death via necrosis by 4.5-fold compared to the control6.

The study by Raveesha et al. (2022) details the rational design and evaluation of novel thiazole derivatives as potential anticancer agents, utilizing a combined synthetic and computational approach. The researchers report the successful physical synthesis of a new series of thiazole-based compounds. As is standard for validating novel multi-target directed ligands or heterocyclic hybrids, the structural integrity and purity of the synthesized molecules were confirmed through spectral characterization techniques, which typically include FT-IR, 1H NMR 13C NMR, and high-resolution mass spectrometry. A significant focus of the paper is the application of DFT to evaluate the chemical descriptors of the synthesized derivatives. This computational method is used to map the molecular electrostatic potential (MEP) and calculate the frontier molecular orbital energies (HOMO-LUMO gap). These parameters provide critical insights into the electronic distribution, chemical reactivity, and kinetic stability of the thiazole compounds, helping to explain how specific substituents influence the molecule's overall electronic profile. To correlate the electronic and structural properties with potential biological activity, the authors utilized molecular docking simulations. This predictive pharmacological modeling calculates the binding affinities and visualizes the crucial non-covalent interactions such as hydrogen bonding and pi-stacking between the novel thiazole ligands and the active sites of selected cancer-related protein targets. The combination of DFT and molecular docking establishes a theoretical framework that supports the in vitro anticancer potential of these compounds, offering a structure-activity relationship (SAR) rationale for why certain derivatives in the series exhibit stronger binding or reactivity than others7.
The study by Raghu et al. (2023) focuses on the development of novel epidermal growth factor receptor (EGFR) kinase inhibitors by designing hybrid molecules that fuse two privileged pharmacophores: quinazoline and thiazole. The primary objective was to discover highly active multi-target agents capable of combating cancer cell proliferation while addressing the therapeutic resistance often encountered with standard tyrosine kinase inhibitors (TKIs) like erlotinib. The researchers synthesized a series of novel quinazoline-based thiazole derivatives via the cyclocondensation of 7-aminoquinazolin-4-ol derivatives with substituted bromoacetophenones. The structural integrity and purity of the resulting hybrid compounds were rigorously confirmed using standard spectral characterization techniques (1H NMR, 13C NMR, and mass spectrometry). The synthesized compounds were evaluated through dual in vitro assays to determine both their broad cytotoxicity and their specific kinase inhibitory activity. The series was tested against three human cancer cell lines: MCF-7 (breast), HepG2 (liver), and A549 (lung), with normal Vero cells used to assess safety and selectivity. The compounds generally exhibited moderate to highly significant cytotoxicity. Compounds 3a and 3b emerged as the most potent agents in the series, displaying efficacy superior to the control drug erlotinib in several instances. Compound 3a exhibited IC50 values of 2.86 µm (MCF-7), 5.91 µm (HepG2), and 14.79 µm (A549)8 .
|
Compound 3b |


Farghaly Ta et al. sought to synthesize thiazole-based chalcones and subsequently develop rigidified 4-hetarylthiazole derivatives, especially focusing on pyrimidine and fused-pyrimidine rings, to develop new antitumor drugs with high cancer cell specificity. In the synthetic route, the E-isomer chalcone intermediates were obtained by aldol condensation of 4-acetylthiazole with the different aromatic aldehyde . The selected chalcones were then submitted to Michael-type addition and subsequent cyclo condensation with thiourea or 6-aminothiouracil to afford the corresponding rigid pyrimidine-thiones and pyridopyrimidine-thiones. The synthesized compounds were evaluated for their anti-proliferative activity against human hepatic (HepG-2), lung (A549) and breast (MCF-7) cancer cell lines by MTT assays. The most potent compound was the chalcone derivative 3a with 4-methoxyphenyl moiety with IC50 values of 1.56 µM, 1.39 µM and 1.97 µM against HepG-2, A549 and MCF-7 respectively. This showed an efficacy of about 2 folds to that of reference drug Doxorubicin. Moreover, compound 3a had a high selectivity index, with much less toxicity to normal WI-38 lung cells. A crucial SAR finding showed that the rigidification of the chalcone enone linker into pyrimidine or fused-pyrimidine ring systems did not lead to enhanced anti-proliferative activity relative to the parent chalcone structures. Flow cytometry and ELISA assays of A549 cells treated with compound 3a showed significant arrest of the cell cycle in the G2/M phase. The compound induced apoptosis by up-regulation of the pro-apoptotic proteins Bax (10.12 folds), active caspase-3 and p53 and down-regulation of the anti-apoptotic protein Bcl-2 (4.12 folds). The lead compounds were tested for their inhibitory effect against cyclin dependent kinases (CDKs). Results showed a non-selective and potent inhibitory activity against CDK1, CDK2 and CDK4 with IC50 values from 0.78 µM to 1.97 µM. Binding affinities and interactions were investigated by molecular docking on the ATP-binding site of CDK1 enzyme (PDB ID: 4Y72). The active compounds docked securely into the active site, anchored primarily by crucial hydrogen bonds. The thiazole sulfur and 2-methylamino groups interacted directly with the Leu83 residue to establish this primary connection. Additionally, the binding pocket was stabilized by a secondary hydrogen bond with Glu81, alongside a supportive network of hydrophobic interactions involving Ile10, Tyr15, and Val18 9.
THIAZOLE DERIVATIVES AS ANTIMICROBIAL AGENTS
Globally, bacterial infections are becoming more common and are mostly responsible for morbidity and mortality in poorer nations. Additionally, a major issue is the rise in bacteria that have developed multiple drug resistance as a result of antibiotic overuse. Patients with impaired immune systems and those who are susceptible to opportunistic fungal infections are especially vulnerable to this issue10 .
Kartsev et al utilized a molecular hybridization approach to design, synthesize, and evaluate the antimicrobial potential of novel heteroaryl (aryl) thiazole derivatives. The objective was to combine thiazole, phthalazine, and sulfonamide moieties into single hybrid molecules to combat the global rise in antibiotic resistance. The synthesis yielded three structural series comprising nine target compounds through distinct chemical routes: acylation of amines with acid chlorides, nucleophilic substitution, and a unique recyclization of 1-chloromethyl-3,4-dihydroisoquinolines under the action of thioamides (for compound 4). The synthesized compounds were evaluated against a panel of bacterial and fungal strains using a microdilution method to determine minimum inhibitory, bactericidal, and fungicidal concentrations. The derivatives displayed moderate to good antibacterial activity. Compound 4 exhibited the highest antibacterial potency, achieving Minimum Inhibitory Concentrations (MIC) of 0.23–0.70 mg/mL and Minimum Bactericidal Concentrations (MBC) of 0.47–0.94 mg/mL. Across the tested strains, B. cereus was the most susceptible, while E. coli demonstrated the highest resistance. Against resistant strains, compounds 2, 3, and 4 showed greater potency against Methicillin-resistant Staphylococcus aureus (MRSA) than the reference drugs ampicillin and streptomycin. Compound 4 also demonstrated stronger activity against P. aeruginosa than ampicillin. Docking results indicated that the antibacterial activity likely stems from the inhibition of the E. coli MurB enzyme. The compounds displayed favorable free binding energies (-7.02 to -9.96 kcal/mol). Structural analysis revealed that highly active compounds formed crucial hydrogen bonds with the Ser229 residue, which plays a vital role in proton transfer during peptidoglycan synthesis. In silico predictions revealed that all synthesized compounds possessed a bioavailability score of approximately 0.55 alongside favorable drug-likeness profiles. Compounds 1 and 2 yielded the most optimal predictions, achieving high drug-likeness scores (1.03 and 1.09, respectively) without violating any Lipinski, Ghose, Veber, Egan, or Muegge rules11 .

Antimicrobial resistance (AMR) is a critical global public health issue that leads to serious infections, extended hospitalizations, and treatment failures. Sana saffour et al are actively incorporating heterocyclic scaffolds specifically 1,2,4-triazole and 1,3-thiazole rings into new molecules, as these structures are known for their strong antimicrobial and antifungal properties. The researchers successfully synthesized thirteen new chemical compounds. Seven of these compounds feature a 1,3-thiazole ring, while the remaining six contain a 1,2,4-triazole ring. The chemical structures of all final compounds were verified using LC-MS, 1H-NMR, and 13C-NMR spectroscopic techniques. The biological efficacy of these compounds was tested in vitro against twelve distinct bacterial strains and ten fungal species.
Derivative 5a demonstrated the most notable antibacterial activity, exhibiting a specific selectivity toward the Yersinia enterocolitica bacterial strain. In-silico studies revealed that specific thiazole derivatives (compounds 5c, 5d, and 5e) successfully interacted with the active cavity of the lanosterol 14a-demethylase enzyme, which is a major target for antifungal drugs12.

THIAZOLE DERIVATIVES AS ANTI INFLAMMATORY AGENTS
Gajendra kumar et al synthesized a new series of non-steroidal biologically active thiazole- and oxazole-substituted benzothiazole derivatives efficiently by using ultrasound irradiation which drastically reduced reaction times compared with conventional methods. The structural configurations of the synthesized compounds were confirmed by spectroscopic and elemental analysis techniques. The compounds were tested in albino rats, and showed different degrees of pharmacological activity, such as anti-inflammatory, analgesic and free radical scavenging activities. Of the newly synthesized molecules, compound 6 (ethyl substituent) was found to be the most potent, with better therapeutic efficacy than the reference drugs and much lower ulcerogenic liability and good acute toxicity profile13.

Zhen Zhang et al studied about sepsis. Sepsis, driven by systemic inflammatory response syndrome (SIRS), remains a leading cause of global mortality. Given the known anti-inflammatory properties of both 3,5-diaryl-4,5-dihydropyrazole and thiazole derivatives, a novel series of 2-(3,5-diphenyl-4,5-dihydro-1H-pyrazol-1-yl)-4-methylthiazole derivatives was designed and synthesized using a pharmacophore combination strategy. The anti-inflammatory potential of these compounds for sepsis therapy was initially screened via their ability to inhibit lipopolysaccharide (LPS)-induced nitric oxide (NO) release in RAW264.7 macrophages. Structure–activity relationship (SAR) analysis identified compound7 as the optimal candidate, demonstrating superior anti-inflammatory potency compared to indomethacin and dexamethasone. Mechanistically, compound 7 suppressed the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), thereby reducing the production of NO, interleukin-beta (IL-1beta), and tumor necrosis factor-alpha (TNF-alpha) by blocking the MAPKs signaling pathway. Furthermore, in vivo evaluation in C57BL/6J mice revealed that compound 7 significantly mitigated LPS-induced sepsis and minimized multi-organ toxicity, highlighting its potential as a promising therapeutic agent for sepsis14 .
|
coccoco |

Jaismy Jacob et al developed an environmentally safe, cost-effective, multi-component "one-pot" green synthetic method using water as a solvent and tetrabutylammonium fluoride (TBAF) as a catalyst. The synthesis yielded 3 thioureas (4a-c), 13 substituted thiazoles (6a-m), and 5 substituted thiazolidenes (6n-r). Bulky aromatic structural features (a 4-phenyl group and a 5-thiophene-2-carbonyl group) were introduced to selectively target the larger active site pocket of the COX-2 enzyme and block its hydrophobic channel. Out of the synthesized series, compound 8 bearing a diphenylamino group and a 4-tolyl group on the thiazole coreemerged as the most potent dual inhibitor. Compound 6l exhibited significant dual inhibitory activity with IC50 values of
Column 1: Data regarding IC50 value of compond 8
|
COX 1 |
5.55 |
|
COX 2 |
0.09 |
|
5LOX |
0.32 |
Compound 8 demonstrated a high selectivity index for COX-2 over COX-1 (SI= 61.66), making it comparable to the commercial drug etoricoxib (SI = 91.28). Selected compounds, including 6l, markedly reduced downstream inflammatory mediators (PGE2 and LTB4) in LPS-challenged RAW 264.7 macrophage cells in a concentration-dependent manner.
In carrageenan-induced paw edema models using male Wistar rats, oral administration of compound 6l resulted in a 60.82% reduction in inflammation after 6 hours. This outperformed the standard drug indomethacin, which achieved a 53.21% reduction. Doses up to 2000mg/kg in rats showed no signs of toxicity or structural changes in liver, kidney, stomach, or intestinal tissues on histopathological examination. Unlike indomethacin (which caused severe mucosal sloughing and ulceration), compound 8 maintained normal gastric mucosa histology, verifying its superior gastrointestinal safety profile. Real-time PCR analysis of inflamed rat paw tissues confirmed that compound 8 significantly down-regulated and suppressed the up-regulation of COX-2 and 5-LOX genes while sparing COX-1 expression. Computational modeling supported the biological assays, showing strong protein-ligand interactions. Compound 6l achieved optimal binding scores (COX-2= -7.54 Kcal/mol 5-LOX} = -6.99 Kcal/mol due to hydrogen bonding and hydrophobic cavity fitting15 .

THIAZOLE DERIVATIVES AS ANTICONVULSANT AGENTS
Epilepsy is very dangerous CNS diseases due to hyper synchronous electrical discharge in neurons. Globally, around fifty million people are suffering from this destructive neurological condition and out of these, ninety percent patients are from developing countries. Moreover, the risk of seizure, trauma, hospitalization and mortality is very high in epileptic patients which have very strong negative impact on patient's physical, mental and social health . Although, several newer classes of antiepileptic drugs (AEDs) have emerged in the past 15 years, but complete treatment of epilepsy is still a long way. Moreover, these AEDs are ineffective against the 30% refractory epileptic patients and possess severe side effects such as, neurotoxicity, anemia, hepatic failure etc.
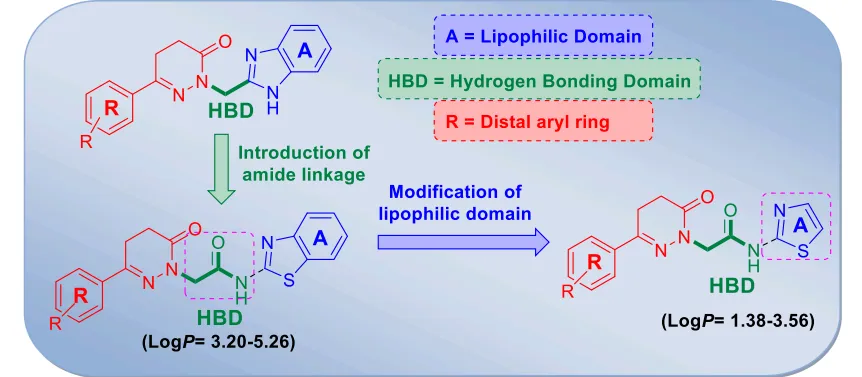
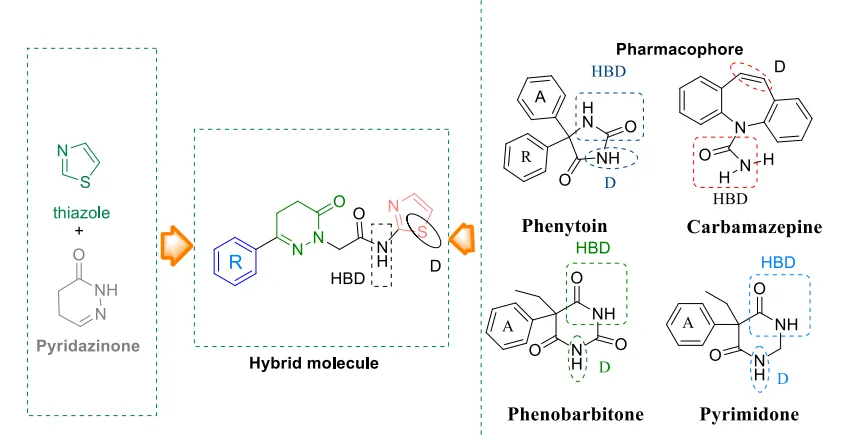
Figure 3: Lead optimization of anticonvulsant
The study aimed to develop newer, safer, and more effective antiepileptic drugs (AEDs) to address the needs of the ~30% of epileptic patients refractory to current medication and to minimize common AED side effects like neurotoxicity and hepatotoxicity. The authors synthesized a series of hybrid molecules connecting a pyridazinone ring and a thiazole moiety via an amide linkage. A series of 18 hybrid analogues (compound -9a to compound -9f) were prepared. All 18 compounds were initially screened intraperitoneally in mice at 30, 100, and 300 mg/kg. Featuring a p-chlorophenyl substitution compound 9f emerged as the most potent analogue. It showed rapid onset and long duration of action, protecting against hind limb tonic extension (HLTE) at a minimal dose of 30 mg/kg in the MES test. Quantitative Phase II evaluation of compound 9f revealed excellent median effective doses ED50of 24.38mg/kg(MES) and 88.23mg/kg (scPTZ). Its Protective Index (PI) was exceptionally high compared to standard drugs 27.30 in MES and 7.54 in scPTZ. Whole-brain GABA estimation in rats treated with compound 9f demonstrated a marked, significant increase in GABA levels (both 2 hours post-acute treatment and after 7 days of chronic oral dosing). Chronic administration resulted in a 1.74-fold increase in brain GABA levels compared to the control, suggesting a GABA-modulatory mechanism. Unlike standard AEDs, a 15-day chronic administration of compound 9f in rats showed no significant alterations in liver enzymes (SGOT, SGPT, Alkaline Phosphatase) or total proteins. Liver tissue staining confirmed a normal histological appearance of the central vein and portal triad area without signs of liver damage. The study successfully optimized pyridazinone analogues into a novel series of hybrid thiazole-pyridazinones. Compound compound 9f stands out as a highly potent, safer anticonvulsant lead candidate that targets the GABAergic system with zero observable neurotoxicity or hepatotoxicity16 .

Davydov et al details the design, synthesis, and evaluation of novel thiopyrano[2,3-d]thiazole derivatives aimed at expanding potential antiepileptic chemical space. Given that a massive proportion of epilepsy cases remain refractory to conventional treatments, discovering small molecules with optimized therapeutic windows is critical. Calculated in silico profiling via SwissADME showed satisfactory overall drug-likeness for the series, with most complying with Lipinski and Veber criteria.
Pharmacological assessment was performed in vivo utilizing the subcutaneous pentylenetetrazole (scPTZ) model in mice. Out of sixteen tested variants, three derivatives—compounds 6, 8, and 10—demonstrated a pronounced anticonvulsant effect. These hit molecules notably prolonged the latency of clonic seizures, minimized seizure frequencies, and diminished animal mortality. Remarkably, compound 10 generated a therapeutic output equivalent to the benchmark drug sodium valproate.
In vitro cytotoxicity assays validated that all three hits possess exceptional safety profiles, failing to reach IC50thresholds against human lymphocytes or HEK293 cell lines up to 100muM Furthermore, molecular docking and dynamic simulations targeting the GABAA receptor demonstrated that the top compound 10 creates a high-affinity, stably bound target complex matching the performance of diazepam17 .

THIAZOLE DERIVATIVES AS ANTIDIABETIC AGENTS
Diabetes mellitus is a chronic, non-infectious metabolic disorder characterised by dangerously high blood glucose levels. A primary therapeutic strategy for managing Type 2 diabetes involves inhibiting carbohydrase enzymes, specifically α-amylase and α-glucosidase. Blocking these enzymes delays the breakdown of carbohydrates and the subsequent absorption of glucose. Because existing oral medications can cause adverse side effects, scientists are actively searching for new, safer therapeutic compounds. Both benzimidazole and thiazole structural scaffolds are already widely recognized for their diverse pharmacological properties. Building on the known benefits of these structures, researchers aimed to synthesize 17 novel hybrid analogs that combine benzimidazole and thiazole rings into single molecules. Their goal was to discover highly potent dual inhibitors of both α-amylase and a-glucosidase. The targeted derivatives, designated as compounds 1 through 17, were synthesized via a straightforward two-step chemical process. Initially, 2-mercapto benzimidazole thiol was reacted with 4-nitro-substituted phenacyl bromide to form an intermediate compound. Subsequently, this intermediate underwent a one-pot reaction with thiosemicarbazide and various substituted 2-bromoacetophenones to produce the final hybrid molecules. The precise chemical structures of these newly formed compounds were rigorously verified using advanced NMR and HREI-MS spectroscopic techniques.To assess their biological activity, the synthesized analogs were tested for enzyme inhibition in vitro, using the established diabetes drug acarbose as a comparative standard. The findings were highly encouraging, as the analogs displayed a wide range of inhibitory potentials. Notably, seven specific derivatives (analogs 1, 2, 3, 4, 5, 10, and 15) demonstrated significantly stronger inhibitory capabilities than standard acarbose. Analog 3, distinguished by a para-fluoro substitution, emerged as the single most potent inhibitor in the entire series.The study included a detailed structure-activity relationship (SAR) analysis to determine how different chemical substitutions influenced enzyme inhibition. Compounds featuring smaller substituents, such as fluorine and chlorine, or those containing groups capable of hydrogen bonding (like hydroxyls), showed remarkably increased inhibitory strength.Conversely, adding bulky substituents like bromine, or non-hydrogen-bonding groups like methyls, resulted in lowered enzyme inhibition. The exact placement of these substituent groups (whether ortho, meta, or para) on the phenyl rings drastically altered the overall effectiveness of the compounds. Finally, molecular docking studies were utilized to visualize exactly how these synthetic compounds interact with their target enzymes. Computer simulations revealed that the most potent analogs successfully adopted conformations allowing for crucial interactions, including hydrogen bonding, directly within the active catalytic cavities of both α-amylase and α-glucosidase18.
Bhat AA et al intelligently fuse two highly versatile five-membered heterocyclic scaffolds the nitrogen-containing pyrrolidine and the sulfur- and nitrogen-containing thiazole into a single hybrid molecule. The central premise of the research is that the molecular hybridization of these specific pharmacophores can yield novel conjugates with significantly enhanced binding affinities and superior pharmacological profiles compared to traditional single-target therapies. The investigative core of this study heavily relies on rigorous in silico molecular docking techniques to predict how these designed conjugates interact with three critical enzymatic targets: α-amylase, α-glucosidase, and dipeptidyl peptidase IV (DPP-IV). By designing ligands capable of simultaneously targeting these specific proteins, the researchers aim to suppress postprandial carbohydrate digestion while also enhancing incretin stability, creating a dual-action mechanism that is highly effective for maintaining glucose homeostasis. The authors successfully delineate a comprehensive structure-activity relationship (SAR) for the designed series. Through detailed docking simulations, they meticulously map the stabilizing forces specifically critical hydrogen bonding, π–π stacking, and π–alkyl interactions anchoring the ligands within the receptor active sites. The computational data compellingly suggests that several of the proposed derivatives possess binding affinities that easily surpass established clinical standards, such as acarbose and vildagliptin. From a robust medicinal chemistry perspective, the review successfully highlights the sheer efficiency of integrating advanced computational modeling into early-stage rational drug design. By prioritizing high-resolution docking profiles, the authors efficiently screen a large library of hypothesized compounds, firmly establishing theoretical efficacy prior to engaging in resource-intensive laboratory bench synthesis and exhaustive in vitro or in vivo testing. This streamlined methodology successfully minimizes the empirical trial-and-error commonly associated with synthesizing complex heterocyclic hybrids. However, it is important to acknowledge that the study is fundamentally anchored in computational predictions. While the energetic docking scores are incredibly promising and the structural rationale for hybridizing pyrrolidine and thiazole rings is mechanistically sound, the definitive validation of these conjugates absolutely requires rigorous empirical in vitro enzymatic assays. Follow-up pharmacokinetic evaluations will also be essential to definitively confirm their physiological bioavailability, metabolic stability, and overall lack of clinical cytotoxicity. Ultimately, this comprehensive computational research article perfectly serves as an innovative, brilliant blueprint for computationally designing multi-target antidiabetic agents19 .
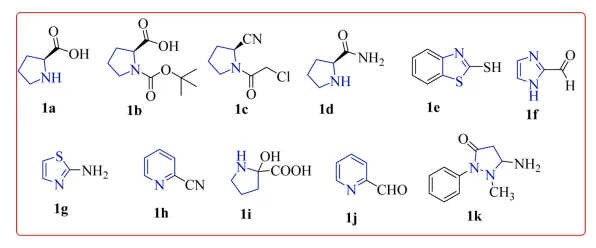
Figure 4. Pyrrolidine and thiazole ring containing derivatives with varying substitutions19 .
THIAZOLE DERIVATIVES AS ANTIVIRAL AGENTS
Gürsoy et al. presents a robust molecular hybridization strategy aimed at overcoming the escalating global crisis of antimicrobial and antiviral resistance. The study leverages the versatile imidazo[2,1-b]thiazole fused bicyclic system—a privileged nitrogen- and sulfur-rich heterocyclic core highly valued for its broad-spectrum pharmacological potential.
The authors construct a well-conceived synthetic pipeline, first generating a series of acyl-hydrazone intermediates. These open-chain precursors are subsequently subjected to a cyclocondensation reaction to yield complex, highly rigid spirothiazolidinone derivatives. From a medicinal chemistry perspective, this conversion to a spirocyclic system is highly strategic; the introduced three-dimensional steric bulk often enhances target specificity, improves metabolic stability, and limits off-target toxicity compared to flatter, planar aromatic systems.
Following synthesis and rigorous structural characterization through IR and NMR spectroscopy, the derivatives underwent comprehensive in vitro screening. The antimycobacterial activity was evaluated against Mycobacterium tuberculosis H37Rv utilizing the Microplate Alamar Blue Assay (MABA). Remarkably, the spirothiazolidinone derivatives—specifically compounds with hydroxy substitution significant antitubercular efficacy, underscoring the vital role of the spiro-linkage for this specific bioactivity. Simultaneously, the antiviral screening across a diverse panel of mammalian cell lines revealed impressive results. Compound 11a emerged as a potent inhibitor of the Coxsackie B4 virus, while compound 11b displayed notable efficacy against both Feline coronavirus and Feline herpesvirus. The structure-activity relationship (SAR) clearly dictates that the spirothiazolidinone architecture significantly outperforms the precursor hydrazones in terms of biological target engagement.


Critically reviewing the study, the authors deliver a highly efficient, synthetically accessible pathway to structurally diverse, dual-action therapeutic leads. The empirical in vitro validation is thorough and encouraging. However, a notable limitation of the paper is the absence of computational pharmacology. The integration of in silico molecular docking studies evaluating binding energies and spatial interactions with specific viral proteases or mycobacterial cell wall enzymes would have provided invaluable mechanistic insights to rationalize the observed SAR. Furthermore, predictive pharmacokinetic (ADMET) profiling would help assess the overall drug-likeness and bioavailability of these novel spiro-conjugates.
Ultimately, this article provides a strong empirical foundation for infectious disease drug discovery. It successfully validates the imidazo[2,1-b]thiazole spiro-derivatives as highly promising lead scaffolds, requiring only targeted computational optimization and in vivo pharmacokinetic validation to advance toward clinical viability20.
Foot-and-Mouth Disease Virus (FMDV) is a highly contagious pathogen that causes devastating economic losses in the global livestock industry. Developing effective and safe antiviral agents against FMDV is a pressing priority in veterinary medicinal chemistry. In this study, Ola A. Abu Ali, Yousry A. Ammar, Moustafa S. Abusaif, and Ahmed Ragab address this challenge by designing a novel class of therapeutic agents: thiazole-tethered adamantane derivatives. Because adamantane’s unique, bulky three-dimensional structure allows for the precise spatial positioning of drug pharmacophores, combining it with biologically active thiazole rings represents a highly rational approach to drug design. The researchers successfully synthesized a new series of thiazolo-adamantane hybrids in good yields. The synthetic route involved treating the cyanoacetamide of adamantane amine with phenyl isocyanate, alongside α-halogenated nitriles and α-halogenated carbonyl compounds. To ensure the integrity of the synthesized library, the structural identities of the new derivatives were rigorously confirmed using elemental analysis and a comprehensive suite of spectroscopic techniques, including FT-IR, NMR, and mass spectrometry. The core of the study involved testing the new compounds in vitro against FMDV serotype O Pan Asia using baby hamster kidney (BHK-21) cells. Establishing a strong safety profile is critical for antivirals; thus, cytotoxicity (CC50) was first assessed. Key derivatives (compounds 5, 6, 7, and 8) exhibited CC50 values ranging from 1000 to 3000 µg/mL. Compared to the standard adamantane-based antiviral drug amantadine (CC50 = 3000 µg/mL), these compounds proved to be remarkably safe at the cellular level. During antiviral screening, the derivatives successfully reduced viral titers in a concentration-dependent manner, showcasing high inhibitory efficacy. To decode the underlying mechanisms of these findings, the authors detailed the Structure-Activity Relationship (SAR) of the derivatives. Furthermore, they conducted in-silico molecular docking simulations for the most potent molecules, derivatives 5 and 6. The docking studies demonstrated profound binding affinities within the active sites of two vital viral targets: FMDV 3C Protease (PDB: 5HM2) and RNA Polymerase (PDB: 1U09). The compounds anchored tightly into these target sites through complex interactions, including hydrogen bonding, arene-cation bonding, and hydrophobic contacts. This paper presents a cohesive and well-executed workflow from initial structural design to biological and computational validation. By demonstrating both high antiviral efficacy and low cytotoxicity, the authors successfully establish these novel thiazole-adamantane derivatives as highly promising lead candidates for future FMDV therapeutics21 .
CONCLUSION
Thiazole derivatives have long been recognized as the most versatile and privileged scaffolds in contemporary medicinal chemistry. These heterocycles possess a unique five-membered aromatic ring that contains both an electron-donating sulfur atom and an electron-accepting nitrogen atom, giving them a strong and highly tunable base for rational drug design . In the past decade, molecular hybridization approaches have been extensively employed by scientists to improve the pharmacological potential of thiazoles. Scientists have successfully synthesized novel multi-target agents by fusion of the thiazole core with other structurally diverse and active moieties such as pyrazole, quinazoline, benzimidazole, and adamantane . Modern drug discovery has been greatly facilitated by advanced computational modeling, including molecular docking and density functional theory (DFT), which enable rapid identification of compounds with improved target specificity, higher binding affinities and reduced toxicity.
FUTURE SCOPES
The contemporary drug discovery has a great potential in the ongoing discovery of thiazole derivatives. Future research should adopt rational design and advanced computational modelling for the development of multitarget-directed ligands (MTDLs) against complex multifactorial diseases such as cancer, diabetes and epilepsy. Integration of nanotechnology could be further employed to improve the bioavailability and targeted delivery of these compounds and to reduce systemic toxicity. Moreover, thiazole chemistry has a lot of potential to solve the growing global problem of antimicrobial and antiviral resistance through novel mechanisms of action. The remarkable chemical flexibility of this scaffold calls for maximum exploitation in order to provide safer, more efficient and patient friendly therapeutics through the adoption of sustainable green chemistry methodologies to improve the accessibility of diverse analogs.
REFERENCES


Blessna Varghese, Sherin Hameed, Thiazole Derivatives: A Decade of Advances in Synthesis, Biological Activities and Drug Discovery, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 2516-2532, https://doi.org/10.5281/zenodo.21338632
 10.5281/zenodo.21338632
10.5281/zenodo.21338632