
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Godavari institute of Pharmacy Kolpa
Background: Ubrogepant and rimegepant are second-generation, orally administered calcitonin gene-related peptide (CGRP) receptor antagonists ("gepants") approved for the acute and/or preventive treatment of migraine. As small-molecule oral therapeutics with defined degradation-sensitive structures, both require robust, stability-indicating analytical methods for routine quality control, yet no validated method for their simultaneous estimation in a combined dosage form has been established in the literature reviewed. Method: A narrative review of the literature (2009-2026) was conducted using PubMed, ScienceDirect, and Google Scholar to evaluate (i) individually reported stability-indicating chromatographic methods for ubrogepant and for rimegepant, and (ii) the Analytical Quality by Design (AQbD) framework — encompassing Analytical Target Profile (ATP) definition, risk assessment, Design of Experiments (DoE), design-space/Method Operable Design Region (MODR) establishment, and ICH Q2(R2)/Q1A(R2)/Q1B-aligned validation and forced degradation — as applied to structurally analogous simultaneous-estimation method-development studies. Results: Individually, a validated stability-indicating RP-HPLC method for ubrogepant achieved LOD/LOQ of 0.02/0.2 µg/mL, recovery of 100.86%, and %RSD below 0.6% for precision, while a stability-indicating RP-HPLC method for rimegepant sulfate (Nurtec ODT) achieved specificity for both assay and impurities using a C18 gradient method at 265 nm. AQbD-based DoE approaches (Box-Behnken, Plackett-Burman, and factorial designs) applied to structurally comparable simultaneous-estimation problems have consistently identified mobile-phase pH, organic-modifier ratio, and flow rate as the critical method parameters governing resolution and peak purity, with several recent AQbD studies additionally incorporating AGREE/GAPI greenness scoring (reported AGREE scores of 0.62-0.87 across cited analogous methods) alongside conventional ICH Q2(R2) validation. Conclusion: The existing single-drug stability-indicating literature for ubrogepant and rimegepant, combined with the well-established AQbD/DoE/greenness methodology demonstrated across structurally comparable simultaneous-estimation studies, together provide a strong evidence-based template for developing a combined, stability-indicating, green AQbD-based RP-HPLC/UPLC method for the simultaneous estimation of these two oral CGRP receptor antagonists — a method that, to the best of the reviewed literature, has not yet been published and represents a clear research gap
Migraine is among the most prevalent and disabling neurological disorders worldwide, and the discovery of calcitonin gene-related peptide (CGRP) as a central mediator of migraine pathophysiology — driving vasodilation, neurogenic inflammation, and nociceptive signaling within the trigeminovascular system — has reshaped migraine pharmacotherapy over the past decade. Small-molecule CGRP receptor antagonists, collectively termed "gepants," represent the most recent oral therapeutic class in this space, offering an alternative to injectable monoclonal antibodies and to triptans (which carry vasoconstrictive contraindications). Ubrogepant (Ubrelvy®) and rimegepant (Nurtec® ODT) are the two most established second-generation oral gepants: ubrogepant is approved as an oral tablet (50 or 100 mg) for the acute treatment of migraine, while rimegepant, formulated as an orally disintegrating tablet (75 mg, oral bioavailability approximately 64%), is unusual among the class in being approved for both acute and preventive treatment.
As with any oral small-molecule drug substance, ensuring the identity, purity, and stability of ubrogepant and rimegepant throughout manufacturing, storage, and shelf-life requires validated, stability-indicating analytical methods capable of resolving the intact drug from its degradation products, process-related impurities, and (where applicable) co-formulated excipients. Individually, stability-indicating RP-HPLC methods have been reported for each drug; however, a validated method for their simultaneous estimation — relevant to combination-product development, comparative quality-control workflows, or research settings evaluating both gepants side by side — does not appear in the literature surveyed for this review, representing a clear gap that Quality by Design (QbD) methodology is well positioned to address.
This review consolidates (i) the individually published stability-indicating analytical literature for ubrogepant and rimegepant, (ii) the pharmacological and physicochemical rationale for a combined estimation method, and (iii) the Analytical Quality by Design (AQbD) framework — risk assessment, Design of Experiments (DoE), design-space/Method Operable Design Region (MODR) development, and ICH-aligned validation — as demonstrated across structurally and analytically comparable simultaneous-estimation method-development studies, to provide an evidence-based foundation for future AQbD-based simultaneous stability-indicating method development for these two oral CGRP receptor antagonists.
1.1 Review Methodology
A structured literature search was conducted across PubMed/MEDLINE, ScienceDirect, and Google Scholar using combinations of the terms "ubrogepant," "rimegepant," "CGRP receptor antagonist," "gepant," "stability-indicating," "RP-HPLC," "Analytical Quality by Design," "AQbD," "Design of Experiments," and "greenness assessment," restricted predominantly to 2009-2026. Given the absence of a published simultaneous-estimation method for ubrogepant and rimegepant at the time of this review, methodologically comparable AQbD-based simultaneous-estimation studies for other drug pairs were additionally reviewed to characterize the applicable framework, risk factors, and validation/greenness benchmarks relevant to future method development for this drug combination.
2. Ubrogepant and Rimegepant: Pharmacological and Analytical Rationale
CGRP receptor antagonists act by competitively blocking CGRP binding at its receptor complex, thereby preventing downstream vasodilation and sensitization of trigeminal nociceptors implicated in migraine pain. Second-generation gepants, including ubrogepant, rimegepant, and atogepant, were developed specifically to overcome the hepatotoxicity that led to discontinuation of the first-generation agent telcagepant, while retaining oral bioavailability and rapid onset of action. Rimegepant has additionally been shown to antagonize CGRP signaling at both the canonical CGRP receptor and the related AMY1 receptor, a pharmacological nuance with potential relevance to degradation-product specificity requirements in analytical method development, since structurally related impurities may retain partial receptor-binding-relevant substructure.
Figure 1. Schematic mechanism of action of oral CGRP receptor antagonists (gepants), illustrating competitive receptor blockade of CGRP-mediated vasodilation and nociceptive signaling relevant to migraine pathophysiology.
From an analytical-development perspective, both drugs present a combination of challenges relevant to stability-indicating method design: both are small, structurally complex heterocyclic molecules with defined process-related impurities (ubrogepant impurities 1 and 2 have been specifically characterized in the validated literature), both are formulated in immediate-release or orally disintegrating tablet matrices requiring excipient-tolerant chromatographic conditions, and both, as CNS-active, chronically administered oral therapeutics, warrant robust forced-degradation characterization to support long-term stability and quality-control programs as their generic and combination-product landscape develops.
3. Individually Reported Stability-Indicating Methods
3.1 Ubrogepant
Nagababu et al. developed and validated a stability-indicating HPLC method for the quantification of ubrogepant and its process-related impurities 1 and 2, using a ProntoSIL ODS C18 (250 × 4.6 mm, 5 µm) column with a phosphate-buffer-based mobile phase. The method demonstrated adequate sensitivity to detect and quantify both impurities, and forced degradation under acid, base, peroxide, thermal, and UV-light stress conditions produced minimal API degradation while efficiently resolving stress-generated degradation products from the parent compound and known impurities, confirming the method's stability-indicating character.
A separate RP-HPLC method development and validation study for ubrogepant reported linearity across 25-150% of nominal concentration (regression coefficient 0.999), LOD and LOQ of 0.02 µg/mL and 0.2 µg/mL respectively, repeatability precision of 0.54% RSD, intermediate precision of 0.33% RSD, and recovery of 100.86%, with the method additionally shown to effectively separate the drug from its degradation products under forced-degradation stress, confirming stability-indicating capability alongside bench-top and refrigerated solution-stability data extending to seven days.
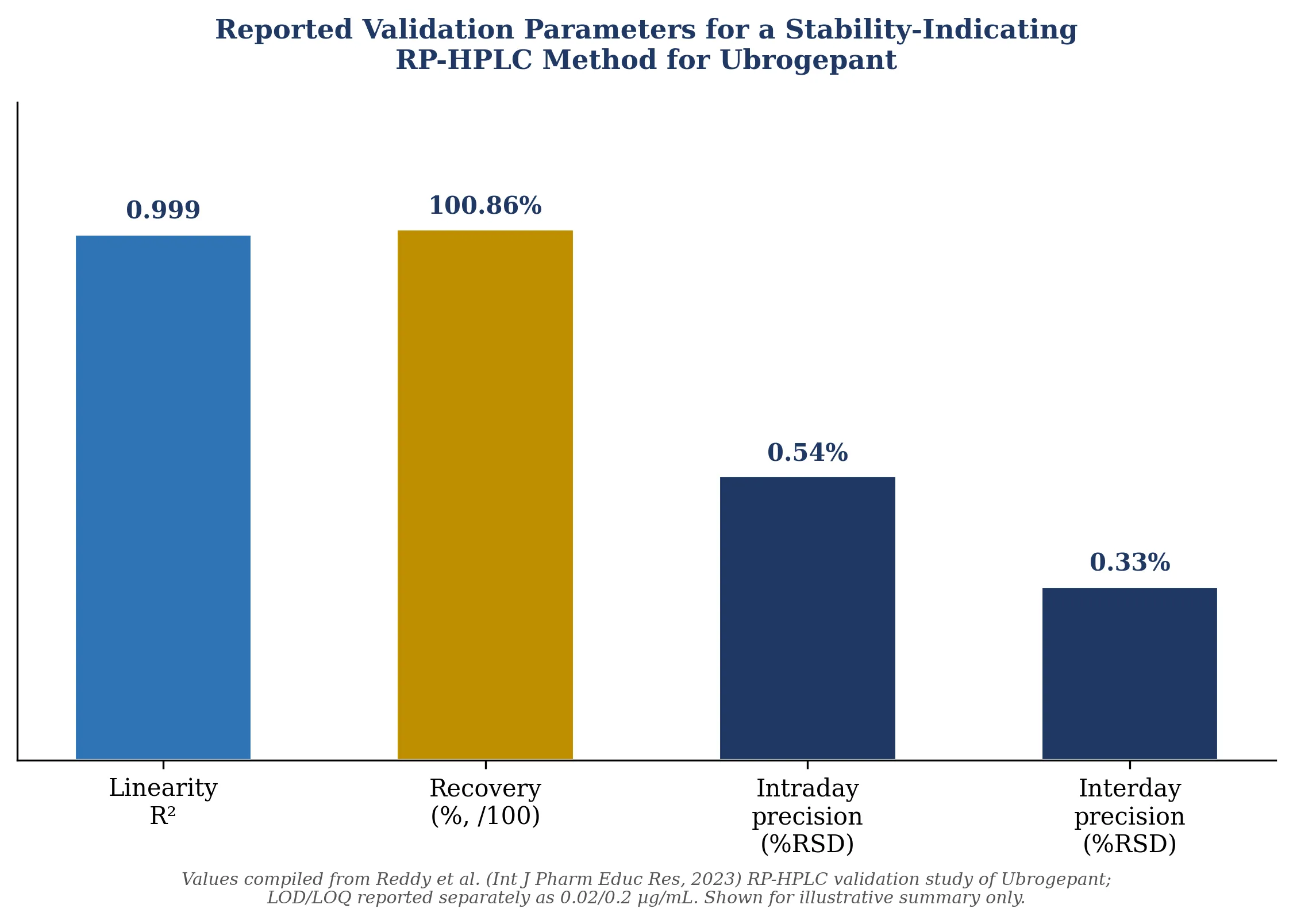
Figure 2. Selected validation parameters reported for a stability-indicating RP-HPLC method for ubrogepant, compiled from the cited primary literature.
3.2 Rimegepant
Kumar et al. validated a stability-indicating RP-HPLC method for the simultaneous estimation of content and impurities of rimegepant sulfate orally disintegrating tablets (Nurtec ODT), using an Agilent Eclipse XDB-C18 (150 × 4.6 mm, 5 µm) column under gradient elution, a flow rate of 1.0 mL/min, column temperature of 30°C, a 25-minute run time, and UV detection at 265 nm. The method was validated for specificity, linearity, accuracy, precision, and robustness for both assay and impurity quantification, establishing a benchmark chromatographic platform (gradient C18 RP-HPLC with UV detection) directly relevant to the design of a combined ubrogepant-rimegepant method, given the shared reversed-phase retention behavior implied by both drugs' comparable log P and heterocyclic structural class.
Table 1. Summary of individually reported stability-indicating method parameters for Ubrogepant and Rimegepant.
|
Parameter |
Ubrogepant (RP-HPLC) |
Rimegepant sulfate (RP-HPLC, Nurtec ODT) |
|
Column |
ProntoSIL ODS C18, 250 × 4.6 mm, 5 µm |
Agilent Eclipse XDB-C18, 150 × 4.6 mm, 5 µm |
|
Elution mode |
Isocratic (phosphate buffer-based) |
Gradient |
|
Detection wavelength |
Not specified in available abstract |
265 nm |
|
Run time |
Not specified in available abstract |
25 min |
|
Linearity |
25-150% of nominal; r = 0.999 |
Reported linear (specific range not in available abstract) |
|
LOD / LOQ |
0.02 / 0.2 µg/mL |
Not specified in available abstract |
|
Precision (%RSD) |
0.54 (repeatability), 0.33 (intermediate) |
Within ICH acceptance (specific values not in available abstract) |
|
Recovery |
100.86% |
Within ICH acceptance (specific values not in available abstract) |
|
Forced degradation stresses applied |
Acid, base, peroxide, thermal, UV light |
Stability-indicating; specific stress conditions not detailed in available abstract |
4. The Analytical Quality by Design (AQbD) Framework
Analytical Quality by Design applies the same risk-based, systematic development philosophy codified for drug product manufacturing under ICH Q8(R2) to analytical method development itself. The process begins with definition of an Analytical Target Profile (ATP) — the method's required specificity, linearity, accuracy, precision, and robustness characteristics — followed by risk assessment (typically Ishikawa/fishbone diagrams and/or Failure Mode and Effects Analysis) to identify Critical Method Parameters (CMPs, e.g., mobile-phase pH, organic-modifier ratio, flow rate, column temperature) likely to influence Critical Method Attributes/Critical Analytical Attributes (CMAs/CAAs) such as resolution, tailing factor, and peak purity.
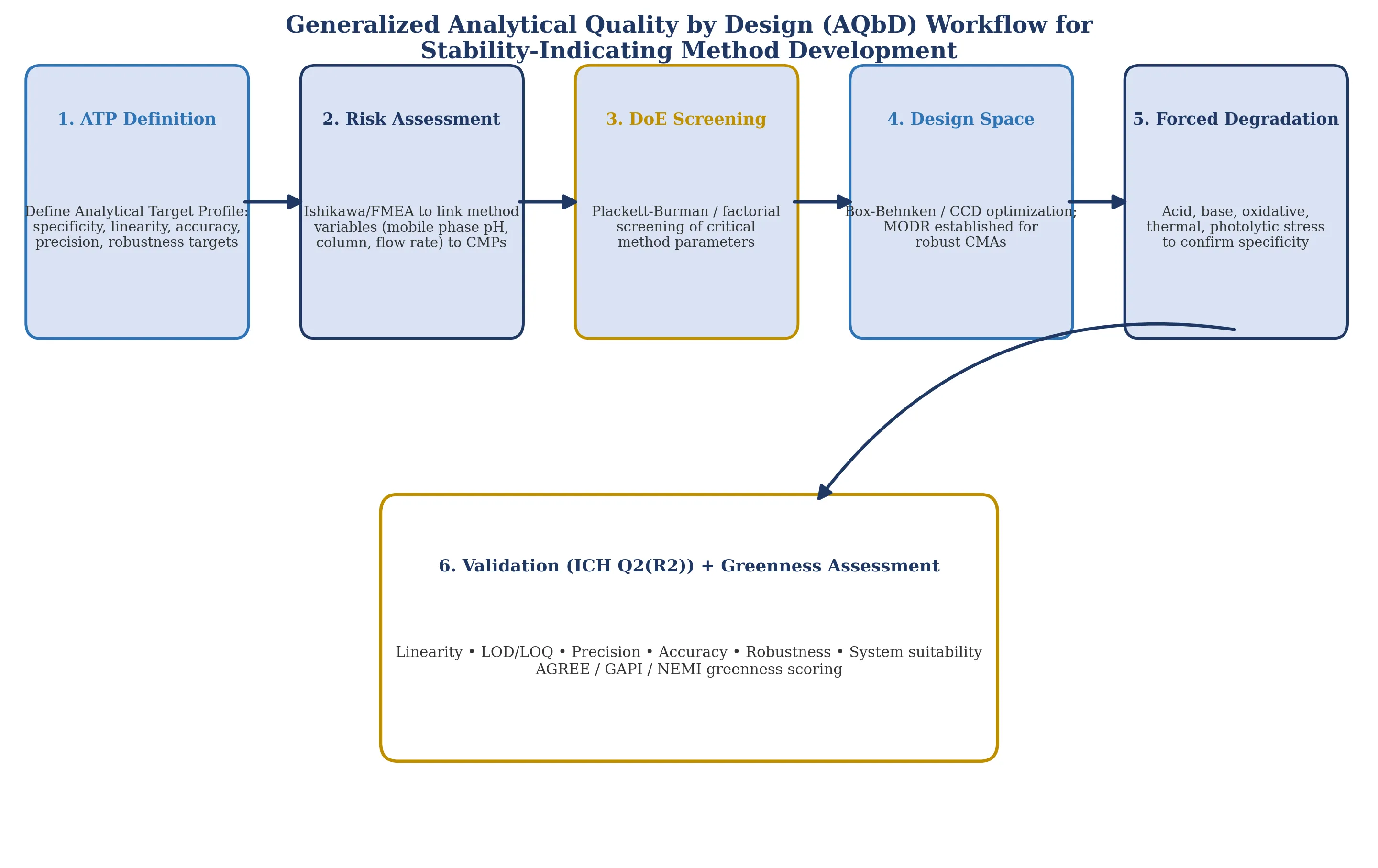
Figure 3. Generalized Analytical Quality by Design (AQbD) workflow for stability-indicating method development, from Analytical Target Profile (ATP) definition through risk assessment, Design of Experiments (DoE) screening, design-space/Method Operable Design Region (MODR) establishment, forced degradation, and ICH Q2(R2)-aligned validation with greenness assessment.
Design of Experiments (DoE) — commonly Plackett-Burman designs for initial screening of multiple CMPs, followed by Box-Behnken or central composite designs for detailed optimization — is then used to map the relationship between CMPs and CMAs, culminating in a defined design space or Method Operable Design Region (MODR): the multidimensional combination of method parameters within which specificity, resolution, and quantitative performance are assured. Across the AQbD-based simultaneous-estimation studies reviewed here (spanning aspirin-lansoprazole, formoterol-aclidinium, tenofovir alafenamide-emtricitabine, escitalopram-clonazepam, deferasirox-deferiprone, and cinnarizine-domperidone combinations), mobile-phase pH, buffer-to-organic-modifier ratio, and flow rate recur as the most consistently identified critical parameters governing resolution and peak-purity outcomes, providing a directly transferable starting risk-assessment framework for a future ubrogepant-rimegepant method.
4.1 Forced Degradation and ICH-Aligned Validation
Consistent with ICH Q1A(R2) (stability testing of new drug substances and products) and ICH Q1B (photostability testing), stability-indicating method validation requires forced degradation under acid, base, oxidative, thermal, and photolytic stress conditions to demonstrate that the method can resolve the intact API from degradation products without interference — a requirement already individually satisfied for ubrogepant (acid, base, peroxide, thermal, and UV stress, per Nagababu et al.) and reported as met for rimegepant sulfate, though with less granular per-stress detail available in the reviewed abstract. Subsequent validation per ICH Q2(R2) requires demonstration of specificity, linearity and range, accuracy, precision (repeatability and intermediate precision), detection and quantitation limits, and robustness — all systematically addressed within an AQbD framework by mapping each validation parameter back to the design space established during DoE optimization, rather than validating a single empirically fixed method.
4.2 Greenness Assessment as an AQbD Extension
A growing number of recent AQbD-based simultaneous-estimation studies additionally incorporate green analytical chemistry (GAC) assessment alongside conventional validation, reflecting the pharmaceutical analytical community's increasing expectation — consistent with the broader industry shift toward sustainable methods — that a method be evaluated not only for accuracy and robustness but also for environmental impact. Commonly applied tools include the Analytical GREEnness metric (AGREE), the Green Analytical Procedure Index (GAPI), and the National Environmental Methods Index (NEMI), which collectively assess solvent hazard, energy consumption, waste generation, and sample/reagent volume. Across the cited AQbD-based comparator studies, reported AGREE scores for optimized RP-HPLC/HPTLC methods ranged from approximately 0.62 to 0.87 (on a 0-1 scale, higher indicating greater greenness), with several studies reporting favorable GAPI pictogram profiles in parallel, illustrating that AQbD-derived design spaces can be deliberately constrained toward greener solvent systems (e.g., reduced acetonitrile/methanol content, aqueous-favoring mobile phases) without compromising validated performance.
Table 2. Critical Method Parameters (CMPs) and greenness benchmarks reported across AQbD-based simultaneous-estimation RP-HPLC studies structurally/analytically comparable to a prospective ubrogepant-rimegepant method.
|
Comparator drug pair / method |
DoE tool |
CMPs identified |
Reported greenness (if assessed) |
|
Aspirin + Lansoprazole |
Central composite / QbD optimization |
pH, initial %B, initial hold time |
Not assessed (pre-dates common greenness reporting) |
|
Formoterol fumarate + Aclidinium bromide |
Design-Expert optimization |
Buffer pH, mobile-phase ratio, flow rate |
Not assessed |
|
Tenofovir alafenamide + Emtricitabine |
3×2 factorial design |
Methanol:formic acid ratio, flow rate |
Not assessed in available abstract |
|
Escitalopram + Clonazepam |
Box-Behnken Design |
Mobile-phase composition, flow rate, wavelength |
Not assessed |
|
Deferasirox + Deferiprone |
Plackett-Burman + custom design |
5 chromatographic parameters screened, then 2-level 3-factor optimization |
8 greenness tools applied (NEMI, AMVI, AMGS, HPLC-EAT, GAPI, AGREE, etc.) |
|
Cinnarizine + Domperidone |
AQbD + fluorimetric detection |
Mobile phase, detection parameters |
GAPI, NEMI, AGREE, Eco-Scale applied |
|
Rivaroxaban (single drug, representative) |
AQbD framework |
Column, mobile-phase pH, flow rate, temperature |
AGREE 0.62; AES 79%; multiple tools applied |
5. Proposed AQbD Framework for a Combined Ubrogepant-Rimegepant Method
Synthesizing the individually validated ubrogepant and rimegepant methods with the AQbD/DoE methodology demonstrated across the comparator simultaneous-estimation literature, a coherent development pathway for a combined method can be outlined. The Analytical Target Profile would specify simultaneous resolution of ubrogepant, rimegepant, and their respective known/process-related impurities and degradation products within a practical run time (informed by the 25-minute rimegepant benchmark as an upper bound, with UPLC sub-2 µm particle technology offering a plausible route to substantial run-time reduction, as demonstrated in comparator stability-indicating UPLC applications for structurally unrelated actives). Risk assessment would reasonably prioritize mobile-phase pH and buffer-to-organic-modifier ratio as the highest-risk CMPs, given their consistent identification across the comparator literature and the differing polarity/ionization behavior implied by ubrogepant's and rimegepant's distinct heterocyclic scaffolds. A Plackett-Burman screening design across candidate CMPs (column chemistry, mobile-phase pH, flow rate, column temperature, gradient slope), followed by Box-Behnken or central composite optimization of the highest-risk factors, would be expected — by direct analogy to the comparator studies reviewed — to establish a robust design space/MODR, with forced degradation (acid, base, oxidative, thermal, photolytic, per ICH Q1A(R2)/Q1B) and full ICH Q2(R2) validation confirming stability-indicating specificity, and AGREE/GAPI assessment providing a quantitative sustainability benchmark consistent with current field expectations.
6. Future Perspectives and Research Gap
This review's most significant finding is methodological rather than comparative: despite both ubrogepant and rimegepant possessing individually validated, stability-indicating RP-HPLC methods, no simultaneous-estimation method for the two drugs was identified in the literature surveyed. Given that both drugs belong to the same pharmacological class, share the acute-migraine-treatment indication, and are structurally amenable to reversed-phase C18 separation under UV detection (as independently demonstrated), this represents a clearly defined and directly addressable research opportunity. Priority directions include: (i) formal AQbD-based development of a combined RP-HPLC or RP-UPLC method incorporating both drugs' known impurities and degradation products within a single run; (ii) application of green-chemistry-constrained DoE (as demonstrated in the deferasirox-deferiprone and cinnarizine-domperidone comparator studies) from the outset, rather than as a post-hoc greenness assessment; and (iii) extension of the validated method to LC-MS/MS platforms to support pharmacokinetic and bioequivalence applications, an area of particular relevance given the ongoing clinical interest in gepant pharmacokinetics and combination-therapy strategies noted in the pharmacological literature reviewed.
7. CONCLUSION
Ubrogepant and rimegepant, as second-generation oral CGRP receptor antagonists central to modern migraine pharmacotherapy, each possess individually validated stability-indicating RP-HPLC methods demonstrating strong sensitivity, precision, and forced-degradation specificity. The Analytical Quality by Design framework — comprising Analytical Target Profile definition, structured risk assessment, Design of Experiments-based optimization, design-space/MODR establishment, and ICH Q2(R2)/Q1A(R2)/Q1B-aligned validation, increasingly extended with AGREE/GAPI greenness assessment — has been robustly and repeatedly demonstrated across structurally comparable simultaneous-estimation method-development studies for other drug combinations. The convergence of these two literatures provides a clear, evidence-based template and a well-defined research gap: a combined, AQbD-optimized, stability-indicating, green analytical method for the simultaneous estimation of ubrogepant and rimegepant remains an open and readily addressable opportunity for future analytical method-development research.
Conflict of Interest
The authors declare no conflict of interest.
ACKNOWLEDGMENT
The authors acknowledge [Institution/Department name] for providing the necessary literature access and support for this review.
REFERENCES

Dr. Rahul Solunke*, Kothale Sakshi, A QbD-Based Approach to Stability-Indicating Analytical Method Development for the Simultaneous Estimation of Oral CGRP Receptor Antagonists a Comprehensive Review, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 2337-2346. https://doi.org/10.5281/zenodo.21317933
 10.5281/zenodo.21317933
10.5281/zenodo.21317933