
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Quality Assurance, S. G. S. P. S. Institute of Pharmacy Kaulkhed, Akola
A simple, rapid, accurate, and precise RP-HPLC method was developed and validated for simultaneous estimation of Dutasteride and Silodosin in bulk and tablet dosage form. Chromatographic separation was performed on a C18 column with methanol: water:0.1% OPA 80:20 v/v as mobile phase at 1.0 mL/min flow rate and detection at 225 nm. Retention times were 3.2 min for Silodosin and 5.8 min for Dutasteride. The method showed linearity in the range of 5-30 µg/mL for Silodosin and 1-6 µg/mL for Dutasteride with correlation coefficient >0.999. The %RSD for precision was <2% and mean %recovery was 99.2-101.1%. LOD and LOQ were found to be within limits. The method was validated as per ICH Q2(R1) guidelines for linearity, accuracy, precision, specificity, and robustness. The developed method can be successfully applied for routine quality control analysis of both drugs in combined pharmaceutical formulations.
The primary goal of pharmaceutical analysis is to assure drug quality. It is well known that quality cannot be test into a product; however, well plan testing with suitable methodology and instrumentation can help build quality into a drug product. It is essential to understand potential degradation reaction that may occur in the the formulated product under various stress conditions that might be encountered during storage and inshipment of final package. The combined dosage forms are complex in nature during process of estimation, it is important to confirm that one component does not interfere with estimation of other. There is a plethora of analysis of such formulations without prior separation. For the estimation of multi component formulation, the instrumental techniques, which are commonly employed, are spectrophotometry, GLC, HPTLC and HPLC etc., these methods are based upon the measurement of specific and nonspecific physical properties of the substances.
2. Chromatography2 Chromatography, by classical definition, is a separation process where resolution is achieved by the distribution of the components of a mixture between two phases, a stationary phase and a mobile phase. Those components held preferentially in the stationary phase are retained longer in the system than those that are distributed in the mobile phase. As a consequence solutes are eluted from the system in the order of their increasing distribution coefficients with respect to the stationary phase, a separation is achieved.
2.1. High Performance Liquid Chromatography3, 4The technique of high performance liquid chromatography is so called because of its improved performance in terms of rapidity, specificity, sensitivity, accuracy, convenience, ease of automation and the cost of analysis when compared to classical column chromatography. Advances in column technology, high pressure pumping system and sensitive detectors have transformed liquid column chromatography into a high speed, efficient, accurate and highly resolved method of separation. Modes of Separation in HPLC5There are two modes of separation in HPLC. They are normal phase mode, reversed phase mode.
2.1.1. Normal Phase Mode: The nature of stationary phase is polar and the mobile phase is non-polar. In this Ltechnique, non-polar compounds travel faster and are eluted first because of the lower affinity between the non-polar compounds and the stationary phase. Polar compounds are retained for longer times and take more time to elute because of their higher affinity with the stationary phase. Normal phase mode of separation is not generally used for pharmaceutical applications because most of the drug molecules are polar in nature and hence take longer time to elute.
2.1.2 Reversed Phase Mode: Reversed phase mode is the most popular mode for analytical and preparative separations of compounds of interest in chemical, biological, pharmaceutical, food and biomedical sciences. In this mode, the stationary phase is non-polar hydrophobic packing with octyl or octadecyl functional group bonded to silica gel and the mobile phase is a polar solvent. An aqueous mobile phase allows the use of secondary solute chemical equilibrium (such as ionization control, ion suppression, ion pairing and complexation) to control retention and selectivity. The polar compound gets eluted first in this mode and non-polar compounds are retained for longer time. As most of the drugs and pharmaceuticals are polar in nature, they are not retained for longer time and hence elute faster. The different columns used are octadecyl silane (ODS or C18), octyl silane C8, butyl silane C4 etc., (in the order of increasing polarity of the stationary phase
REVIEW OF LITERATURE
Sadutto et. al, (2025) developed an enantio- and chemo-selective HPLC method for silodosin using an amylose-based chiral stationary phase. The study focused on separation and quantification of enantiomeric impurities, demonstrating the growing importance of stereochemical purity in pharmaceutical analysis. The method provided excellent resolution and sensitivity, indicating its applicability for impurity profiling and regulatory submissions The optimized conditions employed the mixture n-heptaneethanol-diethylamine (70:30:0.1) (v/v/v) as a mobile phase and a temperature of 35 °C. The complete separation of the enantiomers of silodosin and its main impurities was obtained within 12 min. The chromatographic method has been validated according to the International Conference on Harmonization (ICH) guidelines and compared with the method reported in the JP monograph. The standard curve for silodosin exhibited linearity (R2 > 0.999) within the concentration range of 1.13–2500 µg mL−1. The Chiralpak AD-3 has demonstrated a remarkable level of efficiency, enabling the attainment of limits of quantitation for silodosin of 1.13 µg mL−1 (equivalent to 0.057% of a sample solution of 2 mg mL−1) and ranging from 0.48 µg mL−1 to 1.94 µg mL−1 for other impurities.[20]
Mishra et. al, (2024) The study focuses to validate and develop a precise, simple and accurate stability indicating RP-HPLC method for estimation simultaneously of and silodosin and mirabegron in synthetic mixture. The chromatographic separation was achieved by using Shimpack Solar C18 column (250×4.5mm, 5μm) with acetonitrile: 5mM ammonium acetate in ratio of 90:10% v/v as a mobile phase at a constant flow rate of about 1.2mL/min. The development and validation were carried out at detection wavelength of 229nm. We developed a robust RP-HPLC method, validated for linearity, precision, accuracy, specificity, and system suitability. The method demonstrated excellent linearity with correlation coefficient value r2 was nearly 0.998 with linearity range 8-18μg/mL for Silodosin and 24-54μg/mL for mirabegron. LOD and LOQ were found to be lower; hence, the method is sensitive. Percentage recovery was obtained 99.97% and 99.99% for silodosin and mirabegron, respectively. In case of precision, robustness and repeatability, RSD was found to be less than 2. The validated and developed RP-HPLC method offers an efficient and practical approach for the simultaneous quantification of silodosin and mirabegron in pharmaceutical formulations, making it a valuable tool for quality control and pharmaceutical research. [21]
Gupta et. al, (2024) The Reverse phase HPLC method was developed for simultaneous determination of Silodosin and Tadalafil in single analytical method. Chromatographic separation was achieved on a Supelco C8 (150mmx4.6mm, 5µm) column applying an isocratic elution based on premix of potassium phosphate dibasic buffer pH (4.3) and acetonitrile in the ratio of (70:30 v/v) as mobile. Validation parameters specificity, precision and robustness have been observed to be desirable over the concentration ranges of 80-240 µg/ml for Silodosin and 50-150 µg/ml for Tadalafil in accuracy parameter and 128-192 µg/ml for Silodosin and 80-120 µg/ml for Tadalafil in linearity parameter. All the variables have been studied to optimize the chromatographic conditions. The optimized approach verified through validation and confirmed to be intended purpose for the quality control of the mentioned drugs, as per ICH guidelines. For simultaneous quantification of Silodosin and Tadalafil, the developed method was found to be genuinely exact precise, accurate, linear, fast and cost effective. [22]
R. S. Sakhare et. al, (2023) Studied RP-HPLC Method Development And Validation For Simultaneous Estimation of Azelnidipine And Telmisartan In Pharmaceutical Dosage Form By Qbd Approach in this “Quality by Design” (QbD) serves as a bridge between industry and drug regulatory authorities to move towards a scientific, risk based holistic and proactive approach for development of pharmaceutical products. So, the present work describes the development of RP-HPLC method for simultaneous estimation of azelnidipine and telmisartan in pharmaceutical dosage form by QbD approach. The Box-Behnken design was used for screening where the effect of flow rate, % organic phase and temperature on retention time, resolution, number of theoretical plates (NTP) and symmetry factor (critical quality attributes) was evaluated. Chromatogram was run through Discovery C18 250 x 4.6 mm, 5m. Mobile phase containing 0.01N Ortho Phosphoric Acid: Acetonitrile taken in the ratio of 53.8:46.2V/V was pumped through column at a flow rate of 0.9 ml/min. The developed method was validated according to guidelines of the International Conference on Harmonization (ICH). Hence, the developed method using QbD approach was better understood that, reduces the time and cost of the analysis. [23]
Andreas Vrachas, et. al, (2022) Studied that Cetrimide (CE) is a quaternary ammonium compound and a cationic surfactant, which can be used as an antiseptic and preservative
in various formulations. Antiseptic solutions of Cetrimide are available in combination with Chlorhexidine Gluconate (CHG) for external use. Chlorhexidine is a biguanide with high microbicidal activity and is widely known as a skin disinfectant. The present work displays the development and validation of an RP-HPLC isocratic method for the simultaneous determination of CE and CHG. The method consists of a Hypersil SAS C1 (4.6 × 250 mm) 5 µm column, with a mobile phase of 85%/15% v/v MeOH-NaH2PO4 ·H2O, aqueous solution. In addition, 0.2% of triethylamine (Et3N) was added to the buffer for the confrontation of peak tailing, and then the pH was adjusted to 3.0 with ortho-phosphoric acid (H3PO4). The flow rate was set at 1 mL/min, and adequate detection was achieved with a diode array detector (PDA) at 205 nm. The method was successfully validated according to ICH guidelines for specificity, linearity, accuracy, precision and stability for sample and standard solutions. In addition, the robustness of the method was evaluated through statistical and graphical analysis, using a fractional factorial experimental design. [24]
Kumaraswamy Gandla, et. al, (2022) An accurate, precise and reproducible RP-HPLC method was developed and validated for the estimation of lisinopril in bulk and pharmaceutical dosage form. The chromatographic separation was carried out on Phenomenex column C18 (250x4.6mm,5m), column by using the mobile phase Acetonitrile: Buffer 0.1M (70:30 % v/v) at a flow rate of 1.0 mL/ min. The detection was carried out at a wave length of 237 nm. The retention time for Lisinopril was found to be 3.444 respectively. The developed method was validated according to ICH guidelines [25]
Samineni R. et. al, (2022), reported that the objective of the present study is to develop simple, accurate, sensitive and economic method for effective quantitative determination of Balofloxacin in active pharmaceutical ingredient. As well as in Pharmaceutical dosage forms by using HPLC. The newly developed method is validated in accordance with the analytical parameters for quantitative estimation of Balofloxacin in pharmaceutical dosage forms as per ICH guidelines. The method was validated for parameters like accuracy, linearity, precision, specificity, ruggedness, robustness, and system suitability. The detection was carried out using UV detector at 249 nm. The solutions were injected a a constant flow rate of 1 ml/min. the linearity range of Balofloxacin was found to be 10-60 g/ml. The values of % RSD are less than 2% indicating accuracy and precision of the method. The percentage recovery varies from 98-102% of Balofloxacin. LOD were found to be 0.210 µg/ml and LOQ found to be 0.637µg/ml for Balofloxacin. [26]
Baile M. et. al, (2021), Studied that Development and validation of analytical method play an essential role in the discovery, development and manufacturing of pharmaceuticals. Every year, number of drugs entered into the market; hence it is mandatory to develop newer analytical methods for such drugs. After the development, it becomes necessary to validate the new analytical method. Method development is the process which proves that the analytical method is acceptable for use. Validation of analytical method gives information about various stages and parameters like accuracy, precision, linearity, Limit Of Detection, Limit Of Quantification, specificity, range and robustness. Validation should be done as per regulatory guidelines such as ICH guidelines. [27]
AIM & OBJECTIVES
AIM:
Analytical Method Development & Validation For the Simultaneous Estimation of Dutasteride and Silodosin In Bulk & It’s Pharmaceutical Formulation RP-HPLC
OBJEVTIVES:
The objective of High-Performance Liquid Chromatography (HPLC) method development is to establish a robust, specific, and reliable analytical procedure for the qualitative and quantitative determination of the target analyte(s) in a given matrix. The method should ensure adequate separation of the analyte from related substances, thereby enabling accurate and reproducible measurement.
To select suitable drug ie Dutasteride & Silodosin and drug formulation
To apply suitable analytical techniques
To optimize the analytical techniques employed
To validate the method as per ICH guidelines
To select drug or drug combinations and to develop analytical methodology for estimation of drug
Plan Of Work
Procurement of pure drug samples of Dutasteride & Silodosin and their marketed formulation in combination. Analysis of Pure sample by reported methods. Trial of the instrumental methods on pure drug samples which includes the following steps
Spectrophotometry
Determination of scanning wavelength
High Performance Liquid Chromatography
Selection of mobile phase.
Selection of column.
Selection of chromatographic conditions.
Linearity range.
System suitability parameters.
The efforts will also be made to develop the most reliable and highly sensitive High Performance Liquid Chromatographic method for the combination, The steps in Method development will as follows-
Analysis of standard laboratory mixture to see feasibility of the proposed methods.
To adopt the selected methods on marketed formulation.
Recovery studies.
Validation of the proposed methods.
Parameter For Validation To Be Studies
Accuracy
Precision
Specificity
Ruggedness
Robustness
Limit of detection
Limit of quantitation
Linearity and range
Drug Profile
5.1 Dutasteride
Structure: -
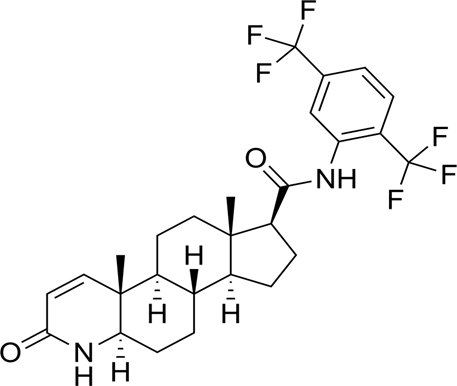
Chemical name: (1S,3aS,3bS,5aR,9aR,9bS,11aS) -N- [2,5-bis (trifluoromethyl)phenyl]-9a,11a-dimethyl-7-oxo-1,2,3,3a,3b,4,5,5a,6,9b,10,11-dodecahydroindeno[5,4-f]quinoline-1-carboxamide
Description
It is white or pale-yellow crystalline powder
Molecular formula: - C27H30F6N2O2
Molecular weight: - 528.539 g/mol
Melting point: - 241-243 0C
Solubility: - Insoluble in water but soluble in organic solvents like ethanol
Category: Dutasteride is synthetic 4-azasteroid an antiandrogenic compound that is used for the treatment of symptomatic benign prostatic hyperplasia (BPH) in adult males by inhibiting 5-alpha reductase. Mechanism of action:-The 5α-reductase is a nuclear-bound steroid intracellular enzyme primarily located in the prostatic stromal cell that converts the analytical information.
MATERIALS & METHODS
MATERIALS
The reliability and scientific validity of an analytical method depend largely on the appropriate selection of materials and the systematic application of well-defined experimental procedures. In HPLC method development, careful consideration of the quality of reagents, instrumentation, and analytical conditions is essential to ensure reproducible, accurate, and precise results. The materials employed must meet suitable purity standards, while the experimental methods should be designed in accordance with established regulatory guidelines to minimize analytical variability and bias The drug used for present investigation was obtained from MG lab Hyderabad as gift sample.
Details of Pure Drug:
Tables of API
|
Sr.No. |
Drug |
Suppliedby |
Quantity |
Purity (Assay) |
|
01 |
Silodosin |
MGLab India |
10g |
99.8 |
|
02 |
Dutasteride |
MGLab India |
10g |
99.2 |
METHODS & PROCEDURE
Identification and characterization of drug
Previous to commenced the experimental work it is necessary to determine the different physical and chemical property of the drug which provide information regarding the purity and nature of drug. This will help in selection of solvents and procedure for the robust and stable analytical method development. Silodosin & Dutasteride
Selection and procurement of drug
Silodosin (SILO) & Dutasteride (DUTA) were selected as model drug candidate for method development and validation. The drugs were kindly gifted from pharmaceutical industry India. The procured drug was analyzed for different physical properties viz. color, odor, melting point, etc.
Physico-chemical characterization The physico-chemical characterization of drug molecule is important with regard to its purity, identification in development and validation of analytical method. The various tools used for characterization of drug molecules include melting point, UV spectroscopy, solubility study, etc.
Solubility Studies As a first step of method development solubility of both drugs was tasted in different solvents to obtain a common solvent which can be used for simultaneous estimation of both drugs in a mixture. Melting point range determination Melting point determination is an essential preformulation and physicochemical characterization parameter that supports HPLC method development. Although melting point analysis is not directly involved in chromatographic separation, it provides valuable information regarding the purity, crystallinity, and thermal behaviour of the drug substance, which indirectly influence method development and optimization. Melting point of Silodosin (SILO) & Dutasteride (DUTA) were determined by placing small amount of sample in capillary tube closed at one end and holds the capillary on melting point apparatus. FT-IR analysis: The IR absorbance spectrum of SILO & DUTA was recorded using FTIR 8400S spectrometer (Shimadzu) over range of 4000 to 400 cm-1. FTIR stands for Fourier Transform Infrared Spectroscopy. The IR spectroscopy theory utilizes the concept that molecules tend to absorb specific frequencies of light that are characteristic of the corresponding structure of the molecules. The energies are reliant on the shape of the molecular surfaces, the associated vibronic coupling, and the mass corresponding to the atoms. It is a technique that uses infrared light to identify chemical properties and molecular structure of materials. FTIR is a modern and preferred method of infrared spectroscopy. The IR absorbance spectrum of SILO & DUTA was recorded using FTIR 8400S spectrometer (Shimadzu) over range of 4000 to 400 cm-1.
UV Spectroscopy Analysis UV spectroscopy plays a pivotal role in the development of high-performance liquid chromatography (HPLC) methods, particularly when UV detection is employed for analyte quantification. Prior UV spectroscopic evaluation of the drug substance provides essential information regarding its absorption characteristics, which directly influence detector wavelength selection and overall method sensitivity. UV spectroscopy also assists in assessing the suitability of mobile phase constituents. Solvents and buffers with significant UV cut-off values can interfere with analyte detection; therefore, UV spectral studies help in selecting mobile phase components with minimal absorbance at the chosen detection wavelength. This step is critical to achieving baseline stability and reducing background noise in HPLC chromatograms. The ultraviolet absorption spectrum of SILO & DUTA were obtained using Shimadzu1800- UV visible spectrophotometer and 1cm quartz cells, over a wavelength range of 400 to 200 nm
RESULT & DISCUSSION
Now a day’s drugs are commonly used clinically and analyst is required to develop suitable method for their analysis. A fixed dose combination containing Silodosin (SILO)& Dutasteride (EZET) is recently available in market as tablet dosage form.Percent purity of above-mentioned drugs were reported by Supplier Company as follows-
1) Silodosin (SILO)- 99.8 %.
2) Dutasteride (DUTA) - 99.2 %
Primary this was not analysed in our study and the % purity stated by the suppliers was taken as standard for comparison studiesThe physico-chemical characterization of drug molecule is important with regard to its purity, identification in development and validation of analytical method. The various tools used for characterization of drug molecules include melting point, UV spectroscopy, solubility study, etc. The solubility study, melting point analysis, UV spectroscopy of the drug was done.
Solubility Study
|
Sr. No |
Solvent / Medium |
SolubilityObservation |
|
1 |
Water |
Practicallyinsoluble |
|
2 |
Methanol |
Freelysoluble |
|
3 |
Acetonitrile (ACN) |
Freelysoluble |
|
4 |
Ethanol |
Soluble |
|
5 |
Dimethylsulfoxide(DMSO) |
Freelysoluble |
SUMMARY & CONCLUSION
L Analytical method development and validation for the simultaneous estimation of Silodosin and Dutasteride in bulk and it’s dosage form by using RP-HPLC has gained the valuable position in the field of analysis due to ease of performance, specificity, sensitivity and the analysis of sample of complex nature. This technique is commonly used for the quantitative estimation of the drugs from their formulation as well as fo studying their metabolites of drugs and their estimation in their biological fluids. This method offers advantages of estimating the constituents for the multicomponant system without prior separation and even nano quantities can be estimated. This technique was employed in the present investigation for simultaneous estimation of Silodosin (SILO) & Dutasteride (DUTA) in tablet dosage form. Careful evaluation of various parameters influencing analysis is an important aspect for the development of analytical method. In order to establish RP-HPLC method the following parameters were studied. HPLC with Inertsil 4.6 (id) x 250 mm column and UV detector was used for the study. The standard and sample solution of SILO and DUTA were prepared in mobile phase. Different pure solvents of varying polarity in different proportions were tried as mobile phase for development of the chromatogram. During selection and optimization of the mobile phase it was observed that the sharpness of the peak is achieved with increasing the proportion of acetonitrile whereas the increased proportion of aqueous resulted in broadening of the peak. The mobile phase that was found to be most suitable was MEOH: Water (60:40) pH 4 and detection wavelength 236 nm was selected for the evaluation of the chromatogram of both drugs. The selection of the wavelength was based on the λ max obtained by scanning of standard laboratory mixture. This system gave good resolution and optimum retention time with appropriate tailing factor (<2). The mean values of system suitability test result are depicted in Table below.
|
Sr.no. |
Parameter |
SILO |
DUTA |
|
1. |
Peakarea |
639535.94 |
108213.16 |
|
2. |
Retentiontime(min) |
4.527 |
7.048 |
|
3. |
Asymmetry |
1.100 |
1.186 |
|
4. |
Efficiency |
114729.55 |
239595.59 |
CONCLUSION
The present study was successfully undertaken to develop and validate a simple, sensitive, precise, and accurate RP-HPLC method for the simultaneous estimation of Silodosin and Dutasteride in bulk and pharmaceutical dosage forms. The primary objective was to establish a reliable analytical procedure that can be effectively employed for routine quality control analysis. During method development, several chromatographic conditions were carefully optimized, including selection of mobile phase composition, pH, flow rate, and detection wavelength. Among the various trials, a mobile phase with pH adjusted to 4.0 was found to be most suitable, as it provided well-resolved peaks with good symmetry and acceptable retention times for both Silodosin and Dutasteride. Detection at 236 nm was selected based on the UV absorption maxima of both drugs, ensuring adequate sensitivity and minimal interference from excipients or other components. The developed method demonstrated excellent chromatographic performance, with clear separation between the two analytes, indicating high specificity. There was no significant interference observed from blank or placebo, confirming that the method is selective for the intended analytes. System suitability parameters such as theoretical plates, tailing factor, and resolution were found to be within acceptable limits, indicating the efficiency and reliability of the chromatographic system. The method was validated in accordance with ICH guidelines, and all validation parameters met the required acceptance criteria. Linearity studies revealed direct and proportional relationship between peak area and concentration over the selected range for both drugs, with correlation coefficients (R²) close to 1, indicating excellent linearity. Precision of the method was confirmed through repeatability and intermediate precision studies, where %RSD values were found to be less than 2%, demonstrating the reproducibility of the method. Accuracy of the method was evaluated using recovery studies at different concentration levels, and the percentage recovery for both Silodosin and Dutasteride was found to be within acceptable limits (98–102%), confirming that the method is accurate and free from systematic errors. The robustness of the method was assessed by making small deliberate changes in chromatographic conditions such as pH, flow rate, and wavelength. The results indicated that the method remained unaffected by these minor variations, highlighting its reliability during normal usage. Furthermore, th method was found to be sensitive, as indicated by low values of limit of detection (LOD) and limit of quantification (LOQ), enabling the detection and quantification of even small amounts of the drugs. The developed method also proved to be rapid, with shorter run times, making it suitable for high-throughput analysis in pharmaceutical laboratories. The applicability of the method was successfully demonstrated by analyzing marketed pharmaceutical dosage forms, where the assay results were found to be within acceptable limits, confirming the suitability of the method for routine quality control analysis. The simplicity of sample preparation and the use of commonly available solvents further enhance the practicality of the method. The developed RP-HPLC method is simple, economical, precise, accurate, robust, and specific for the simultaneous estimation of Silodosin and Dutasteride. The method complies with all validation requirements as per ICH guidelines and can be effectively applied for routine analysis, quality control testing, and stability studies of these drugs in bulk and pharmaceutical dosage forms. This method can also serve as a reliable analytical tool for future research and formulation development involving these compounds.
REFERENCES

Rohan Waghode*, Nusrat Sayyad, Nitin Bhajipale, A Research on Analytical Method Development & Validation for the Simultaneous Estimation of Dutasteride & Silodosin in Bulk & Its Pharmaceutical Formulation RP-HPLC, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7743-7755. https://doi.org/10.5281/zenodo.20433523
 10.5281/zenodo.20433523
10.5281/zenodo.20433523