
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1 M.Tech, Biomedical Data Science, Centre for Life Sciences, Mahindra University, Hyderabad
2 PGDM, Pharmaceutical Management, IIHMR, Banglore, Bengaluru
3 Assistant Professor, Department of Pharmacology, Indus Institute of Pharmacy and Research, Indus University
4 MSc, Transitional Cardiovascular Medicine and Diseases, Allied Health Sciences at Ajeenkya DY Patil University, Pune
5 BSc Clinical Research, Department of Translational and Clinical Research, Jamia Hamdard University, New Delhi
Diabetes mellitus constitutes a global metabolic pandemic, affecting over 537 million adults worldwide as of 2021, with projections reaching 783 million by 2045. The heterogeneous pathophysiology of type 2 diabetes mellitus (T2DM) — encompassing progressive beta-cell dysfunction, hepatic insulin resistance, adipokine dysregulation, gut microbiome perturbations, and neurohormonal imbalance — demands a multidimensional therapeutic framework that transcends single-target pharmacological intervention. This review presents a comprehensive systems pharmacology approach to diabetes management, integrating mechanistic drug therapy across major pharmacological classes (metformin, GLP-1 receptor agonists, SGLT2 inhibitors, DPP-4 inhibitors, sulfonylureas, thiazolidinediones, and insulin analogues) with evidence-based lifestyle modulation (dietary patterns, physical activity, sleep hygiene, and psychosocial interventions). We synthesise molecular network pharmacology, multi-omics data, and cardiovascular outcome trial evidence to construct a personalised, algorithm-driven treatment framework stratified by comorbidity burden, phenotypic subtype, and patient-specific metabolic goals. Key systems-level interactions — including the gut-brain axis, adipose-pancreatic crosstalk, and renal-cardiovascular nexus — are examined to reveal emergent therapeutic targets and polypharmacy risks.
Diabetes mellitus represents one of the most formidable chronic disease burdens of the twenty-first century. The International Diabetes Federation (IDF) Atlas (10th edition, 2021) documented 537 million adults living with diabetes globally, a figure expected to escalate to 783 million by 2045. In South and Southeast Asia alone, India hosts over 101 million individuals with diabetes now surpassing China as the nation with the highest absolute disease burden [1]. The economic ramifications are equally staggering, with global diabetes-attributable expenditure estimated at USD 966 billion in 2021.
The pathophysiology of T2DM once simplistically attributed to impaired insulin secretion and peripheral insulin resistance — has been dramatically reframed over the past two decades. Contemporary understanding embraces an "ominous octet" of pathophysiological mechanisms described by DeFronzo, encompassing hepatic glucose overproduction, reduced incretin effect, hyperglucagonaemia, enhanced renal glucose reabsorption, neurotransmitter dysfunction, accelerated lipolysis, and beta-cell failure superimposed on muscle insulin resistance [2]. This mechanistic complexity renders single-target pharmacotherapy fundamentally insufficient for achieving durable glycaemic control and more critically for preventing the cardiovascular, renal, and neurological complications that drive morbidity and premature mortality in this population [3].
Systems pharmacology the quantitative study of drug action across biological networks, from molecular targets to organ-level and whole-body pharmacodynamic effects provides an epistemological framework ideally suited to diabetes therapeutics [4]. By integrating molecular target biology, polypharmacology, drug-drug interactions, and patient-level physiological parameters into computational and clinical decision models, systems pharmacology enables the rational design of combination regimens that exploit synergistic pathway redundancy while minimising off-target harm [5].
Simultaneously, the primacy of lifestyle intervention in diabetes prevention and management has been unequivocally established. The landmark Diabetes Prevention Program (DPP) demonstrated that intensive lifestyle modification (7% weight reduction and ≥150 minutes/week physical activity) reduced T2DM incidence by 58% in high-risk individuals significantly outperforming metformin monotherapy (31% reduction) [6]. Despite this evidence, lifestyle interventions remain inadequately integrated into pharmacotherapy algorithms in clinical practice, reflecting a compartmentalisation that systems pharmacology can structurally overcome.
This paper presents a comprehensive, evidence-grounded systems pharmacology framework for diabetes management. We review the mechanistic pharmacology of all major antidiabetic drug classes through the lens of network biology, examine the molecular mechanisms underlying lifestyle-induced metabolic modulation, and synthesise these streams into a personalised treatment algorithm that accommodates cardiorenal comorbidities, phenotypic heterogeneity, and patient-centred goals. Our framework incorporates recent cardiovascular outcome trial (CVOT) data, precision medicine genomics, digital health technologies, and behavioural pharmacology to offer a transformative approach to individualised diabetes care.
EPIDEMIOLOGY AND SYSTEMS-LEVEL PATHOPHYSIOLOGY
Global Burden and Phenotypic Heterogeneity
The epidemiological landscape of diabetes encompasses multiple clinical phenotypes with distinct pathophysiological underpinnings. While T2DM constitutes 90–95% of all diabetes cases, it exhibits substantial heterogeneity a fact crystallised by Ahlqvist et al.'s landmark cluster analysis (2018), which identified five distinct T2DM subtypes in 8,980 newly diagnosed patients using hierarchical clustering of six variables: GADA positivity, age at diagnosis, BMI, HbA1c, beta-cell function (HOMA-B), and insulin resistance (HOMA-IR) [7]. These clusters demonstrated markedly different complications profiles and pharmacological responses, presaging the era of precision diabetes medicine.
Type 1 diabetes mellitus (T1DM), characterised by autoimmune beta-cell destruction and absolute insulin dependence, affects approximately 8.4 million globally, with incidence rising 3–4% annually in high-income countries. Latent autoimmune diabetes in adults (LADA) the most common misdiagnosed form shares autoimmune aetiology with T1DM but progresses more slowly, often initially managed as T2DM until insulin dependence manifests [8]. Maturity-onset diabetes of the young (MODY), encompassing at least 14 monogenic subtypes, affects 1–5% of all diabetes patients and may require entirely different pharmacological approaches MODY2 (GCK mutations) rarely requires treatment, while MODY3 (HNF1A) is exquisitely sensitive to sulfonylureas [9].
Gestational diabetes mellitus (GDM) affects 14% of pregnancies globally, with profound implications for both maternal long-term T2DM risk (50% within 10 years) and offspring metabolic programming. The recognition of intra-individual glycaemic variability and continuous glucose monitoring (CGM)-derived metrics such as time-in-range (TIR) has further enriched the epidemiological characterisation of diabetes across its phenotypic spectrum [10].
Molecular Networks in T2DM Pathogenesis
From a systems biology perspective, T2DM pathogenesis can be modelled as a failure of homeostatic network resilience across interconnected physiological modules [11]. The insulin signalling cascade anchored by the insulin receptor substrate (IRS)-PI3K-AKT axis interfaces with nutrient sensing pathways (mTORC1, AMPK), lipid metabolism networks (peroxisome proliferator-activated receptors), and inflammatory signalling (IKKβ-NF-κB, JNK) in ways that create self-reinforcing pathological loops. Lipid overload in hepatocytes activates diacylglycerol-PKCε signalling, impairing insulin receptor kinase activity and driving hepatic insulin resistance. Simultaneously, ceramide accumulation in skeletal muscle activates PP2A, dephosphorylating and inactivating AKT, while promoting inflammatory cytokine production [12].
The gut-brain axis emerges as a critical regulatory nexus in glucose homeostasis and T2DM pathophysiology. Gut-derived GLP-1, secreted from L-cells in response to nutrient ingestion, activates central and peripheral GLP-1 receptors to suppress appetite, slow gastric emptying, stimulate glucose-dependent insulin secretion, and inhibit glucagon release [13]. Gut microbiota dysbiosis characterised by reduced Akkermansia muciniphila, Faecalibacterium prausnitzii, and Lactobacillus species alongside expansion of Bacteroides and Ruminococcus promotes increased intestinal permeability, lipopolysaccharide translocation, and systemic low-grade inflammation that exacerbates insulin resistance [14]. Short-chain fatty acids (SCFAs) particularly butyrate and propionate produced by gut microbial fermentation of dietary fibre activate free fatty acid receptor 2/3 (FFAR2/3) on L-cells and immune cells, modulating both GLP-1 secretion and inflammatory tone [15].
Adipose tissue dysfunction represents a pivotal node in the T2DM network. Visceral adipose tissue expansion activates macrophage polarisation toward pro-inflammatory M1 phenotype, promoting TNF-α, IL-6, and IL-1β secretion that directly impairs insulin signalling in hepatocytes and myocytes through serine phosphorylation of IRS-1/2 [16]. Adiponectin an insulin-sensitising adipokine secreted exclusively by adipocytes is paradoxically reduced in obesity and T2DM, further impairing hepatic fatty acid oxidation and skeletal muscle glucose uptake via AMPK-dependent mechanisms.
Cardiorenal-Metabolic Syndrome Nexus
The concept of cardiorenal-metabolic (CRM) syndrome recognises the bidirectional, mechanistically linked dysfunction of the cardiovascular system, kidneys, and metabolic organs in T2DM [17]. Hyperglycaemia drives endothelial dysfunction through advanced glycation end-product (AGE) accumulation, polyol pathway activation, PKC-mediated NADPH oxidase upregulation, and impaired nitric oxide bioavailability. These mechanisms converge on accelerated atherosclerosis, left ventricular hypertrophy, and glomerular hyperfiltration establishing a self-perpetuating vascular injury cycle that fundamentally shapes both prognosis and pharmacological target selection [18].
The renin-angiotensin-aldosterone system (RAAS) occupies a central pathophysiological position in CRM syndrome. Hyperactivation of intrarenal RAAS promotes glomerular hypertension, mesangial expansion, and tubular fibrosis, while aldosterone excess drives myocardial fibrosis and sodium retention. ACE inhibitors and ARBs while not primarily antidiabetic form an essential pharmacological layer in diabetic nephropathy management by reducing intraglomerular pressure and albuminuria progression [19]. The recent emergence of finerenone (a non-steroidal mineralocorticoid receptor antagonist) as an agent demonstrably reducing CKD progression and cardiovascular events in diabetic kidney disease (FIDELIO-DKD and FIGARO-DKD trials) exemplifies the systems-level thinking that recognises shared pathophysiological nodes across end-organ compartments [20].
SYSTEMS PHARMACOLOGY OF ANTIDIABETIC AGENTS
Metformin: The Foundational Agent
Metformin (1,1-dimethylbiguanide) has occupied the position of first-line pharmacotherapy in T2DM for over six decades, a tenure unprecedented in metabolic medicine and reflecting its favourable benefit-risk profile, low cost, and pleiotropic mechanisms [21]. The primary mechanism inhibition of mitochondrial complex I (NADH:ubiquinone oxidoreductase) reduces ATP production and triggers AMPK activation, which in turn suppresses hepatic gluconeogenesis by phosphorylating and inactivating CREB-regulated transcription coactivator 2 (CRTC2) [22]. AMPK activation additionally promotes skeletal muscle GLUT4 translocation, enhancing peripheral glucose uptake independently of insulin signalling.
Recent mechanistic investigations have elucidated additional metformin targets of pharmacological significance. Metformin activates lysosomal AMPK through a mechanism dependent on aldolase inhibition and v-ATPase, independent of mitochondrial effects [23]. In the gut, metformin inhibits sodium-glucose transporter 1 (SGLT1) in the intestinal brush border, reducing post-prandial glucose absorption, and modulates the gut microbiome increasing Akkermansia muciniphila abundance and SCFA production effects that may explain the dissociation between portal metformin concentrations and systemic AMPK activation [24].
The cardiovascular benefits of metformin extend beyond glycaemic control. The UKPDS 34 substudy demonstrated 39% reduction in myocardial infarction and 36% reduction in all-cause mortality in overweight T2DM patients on metformin monotherapy, effects exceeding those explained by HbA1c reduction alone [25]. Proposed mechanisms include endothelial protection via eNOS phosphorylation, anti-platelet effects, reduction of vascular smooth muscle proliferation, and direct AMPK-mediated cardioprotection against ischaemia-reperfusion injury.
GLP-1 Receptor Agonists: Incretin-Based Systems Modulation
GLP-1 receptor agonists (GLP-1RAs) represent the most significant pharmacological advance in T2DM management of the past decade, with their clinical utility extending far beyond incretin mimicry to encompass cardiovascular protection, renal benefit, and weight reduction of magnitude approaching bariatric surgery [26]. The GLP-1 receptor a class B G-protein-coupled receptor coupled to Gs, triggering cAMP-PKA-CREB signalling is expressed not only in pancreatic beta-cells but also in cardiac myocytes, vascular endothelium, renal tubular cells, hepatocytes, hypothalamic neurons, and vagal afferents, providing a mechanistic basis for the drug class's diverse systemic effects.
The cardiovascular outcome trials (CVOTs) conducted under FDA guidance have irrevocably established GLP-1RA superiority in high-risk T2DM populations. LEADER (liraglutide, n=9,340) demonstrated 13% reduction in three-point major adverse cardiovascular events (3P-MACE) and 22% reduction in cardiovascular mortality [27]. SUSTAIN-6 (semaglutide, n=3,297) showed 26% MACE reduction driven primarily by non-fatal stroke reduction [28]. REWIND (dulaglutide, n=9,901) demonstrated cardiovascular benefit in a broader, lower-risk population 12% MACE reduction extending the evidence base beyond secondary prevention [29].
At the molecular level, GLP-1RA cardiovascular protection involves multiple mechanisms: direct myocardial GLP-1R activation reducing ischaemia-reperfusion injury, anti-atherosclerotic effects through NF-κB suppression and macrophage foam cell regression, BP reduction via natriuresis and sympatholytic effects, and anti-arrhythmic action through cardiac GLP-1R-mediated ion channel modulation [30]. The hepatic GLP-1R (contentiously present at low expression) and indirect hepatic effects via vagal afferents may contribute to the NAFLD/NASH improvements observed with liraglutide (LEAN trial) and semaglutide (ESSENCE trial, Phase 3) [31].
Semaglutide has achieved further pharmacological milestone status with the approval of oral formulation (Rybelsus, 3–14 mg daily) using the SNAC absorption enhancer technology, and the weekly injectable 2.4 mg semaglutide (Wegovy) for obesity management — demonstrating 14.9% body weight reduction in the STEP-1 trialeffectively blurring the therapeutic distinction between antidiabetic and anti-obesity pharmacology [32].
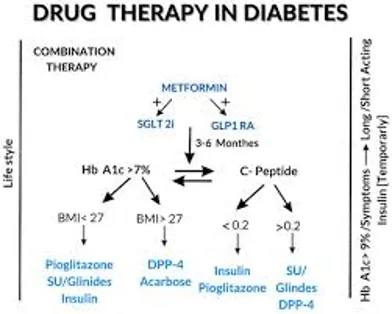
Fig.1 DRUG THERAPY IN DIABETES
SGLT2 Inhibitors: Renal Glycosuria and Cardiorenal Protection
Sodium-glucose cotransporter 2 inhibitors (SGLT2i) represent an insulin-independent therapeutic modality that exploits the kidney's glucose reabsorption capacity as a glycaemic target. Under normoglycaemia, the kidney filters approximately 180g glucose daily, 90% reabsorbed by SGLT2 in the proximal convoluted tubule (S1/S2 segment) and the remainder by SGLT1 in the S3 segment [33]. In T2DM, adaptive SGLT2 upregulation enhances glucose reabsorption as a homeostatic response to hyperglycaemia, paradoxically sustaining elevated glucose levels. SGLT2 inhibition induces glycosuria of 60–90g/day, achieving HbA1c reductions of 0.6–1.0% while simultaneously promoting osmotic diuresis, natriuresis, and caloric deficit [34].
The cardiorenal protective effects of SGLT2 inhibitors revealed through pivotal CVOTs have transformed their pharmacological status from antidiabetic agents to organ-protective cardiovascular medicines [35]. EMPA-REG OUTCOME (empagliflozin, n=7,020) demonstrated 14% reduction in 3P-MACE, 38% reduction in cardiovascular mortality, and 35% reduction in hospitalisation for heart failure findings of such clinical magnitude that they prompted urgent label revisions and guideline updates within months of publication [36]. CREDENCE (canagliflozin) and DAPA-CKD (dapagliflozin) subsequently demonstrated robust renoprotection including among non-diabetic CKD patients in DAPA-CKD establishing SGLT2i as disease-modifying agents in diabetic nephropathy [37].
The mechanistic underpinning of SGLT2i heart failure benefits extends beyond haemodynamic effects of diuresis. The "thrifty substrate hypothesis" proposes that SGLT2 inhibition promotes myocardial metabolic reprogramming from glucose to ketone bodies (β-hydroxybutyrate) a more oxygen-efficient fuel improving cardiac efficiency in the failing heart [38]. Epicardial adipose tissue reduction, sympathetic nervous system inhibition via renal afferent pathways, NLRP3 inflammasome suppression, and autophagy induction represent additional mechanistic contributions to the multifaceted cardiorenal protective phenotype of this drug class [39].
DPP-4 Inhibitors: Incretin Enhancement with Safety Considerations
Dipeptidyl peptidase-4 (DPP-4) inhibitors achieve their antidiabetic effects by preventing the enzymatic degradation of endogenous GLP-1 and GIP (glucose-dependent insulinotropic peptide), extending their half-lives from 1–2 minutes to 12–14 hours. DPP-4 a multifunctional serine exopeptidase expressed on T-lymphocytes (as CD26), epithelial cells, endothelium, and soluble in plasma cleaves a diverse repertoire of substrates beyond incretins, including neuropeptide Y, B-type natriuretic peptide, stromal cell-derived factor-1 (SDF-1), and numerous immunomodulatory peptides [40].
The weight-neutral profile, low hypoglycaemia risk, and once-daily oral administration have established DPP-4 inhibitors as valuable agents in elderly populations and those with hypoglycaemia concerns. However, cardiovascular safety data have been nuanced: SAVOR-TIMI 53 (saxagliptin) demonstrated a significant 27% increase in hospitalisation for heart failure attributed to saxagliptin's broader substrate specificity, including inhibition of SDF-1 degradation and consequent myocardial accumulation of SDF-1 promoting maladaptive cardiac remodelling [41]. TECOS (sitagliptin) and EXAMINE (alogliptin) found no cardiovascular benefit or harm, establishing non-inferiority rather than superiority.
Insulin Analogues: Physiological Replacement and Dose Optimisation
Insulin therapy remains the most potent and universally applicable pharmacological intervention across all stages and phenotypes of diabetes. The pharmacological evolution from animal-derived insulins to recombinant human insulin, and subsequently to analogue insulins with engineered pharmacokinetic profiles, represents one of the most consequential trajectories in pharmaceutical science [42]. Basal insulin analogues — glargine (U-100 and U-300), detemir, and degludec provide sustained, peakless insulin coverage over 24 hours or beyond, reproducing physiological basal insulinaemia and reducing nocturnal hypoglycaemia relative to NPH insulin.
Insulin degludec (Tresiba), with its depot formation of multi-hexameric chains subcutaneously and duration of action exceeding 42 hours, achieves the most stable pharmacokinetic profile among basal insulins and demonstrates 40% lower confirmed hypoglycaemia rates compared to glargine U-100 in the SWITCH trials [43]. Ultra-rapid insulin analogues — faster-acting insulin aspart (Fiasp) with niacinamide addition and URLi (insulin lispro-aabc, Lyumjev) with treprostinil and citrate excipients — exhibit peak action at 40–45 minutes and 4-fold faster initial absorption than conventional rapid analogues, better replicating the first-phase insulin response to meals [44].
Fixed-ratio combination products co-formulating basal insulin with GLP-1RAs — iGlarLixi (insulin glargine/lixisenatide, Soliqua) and IDegLira (insulin degludec/liraglutide, Xultophy) exploit complementary mechanisms to achieve HbA1c reductions of 1.3–1.9% in a single daily injection, while the GLP-1RA component mitigates insulin-induced weight gain and limits hypoglycaemia through glucose-dependent insulin secretion [45].
Emerging Pharmacological Targets
The pharmacological pipeline for diabetes management encompasses a diverse array of emerging targets reflecting systems-level understanding of T2DM pathophysiology. Dual GLP-1/GIP receptor agonists tirzepatide (Mounjaro/Zepbound) activate both GLP-1R and GIPR, the latter potentiating weight loss through central appetite suppression and adipocyte lipid metabolism, achieving unprecedented HbA1c reductions of 2.3% and weight loss of 21% in the SURPASS-5 trial [46]. The approval of tirzepatide across T2DM and obesity indications has created a new pharmacological category, with triple GLP-1/GIP/glucagon receptor agonists (retatrutide, CT-388) in Phase 3 development demonstrating even greater metabolic efficacy.
Finerenone a non-steroidal, selective mineralocorticoid receptor antagonist demonstrated 18% reduction in kidney failure and 13% reduction in cardiovascular events in the combined FIDELIO-DKD and FIGARO-DKD analysis (n=13,026 patients with T2DM and CKD), with a superior safety profile compared to steroidal MRAs (minimal hyperkalaemia, no gynaecomastia) [20]. Its approval for diabetic kidney disease represents a third organ-protective pharmacological pillar alongside RAAS blockade and SGLT2 inhibition in CKD management.
Additional emerging targets include: (1) Glucokinase activators (GKAs) dorzagliatin received approval in China for T2DM, targeting the glucose-sensing glucokinase enzyme in both pancreatic beta-cells and hepatocytes; (2) Ileal bile acid transporter (IBAT) inhibitors — reducing enterohepatic bile acid recirculation and promoting GLP-1 secretion; (3) FGF21 analogues efruxifermin and pegozafermin targeting the FGF21-FGFR1c-KLB axis for NASH and diabetes; and (4) Adipsin/C3aR pathway modulators targeting the complement-mediated pathway of adipose inflammation [47].
Table 1: Comprehensive Pharmacological Profile of Antidiabetic Drug Classes
|
Drug Class |
Mechanism |
HbA1c Reduction |
Key Benefits |
Safety Considerations |
|
Metformin (Biguanides) |
AMPK activation; hepatic glucose output inhibition |
1.0–2.0% |
CV neutral/beneficial; weight neutral; low cost; extensive safety data |
GI intolerance; lactic acidosis (rare); B12 deficiency |
|
Sulfonylureas (e.g., Glipizide) |
Pancreatic beta-cell KATP channel closure; insulin secretagogue |
1.0–2.0% |
Rapid glucose lowering; low cost; oral administration |
Hypoglycaemia risk; weight gain; beta-cell exhaustion |
|
DPP-4 Inhibitors (Gliptins) |
GLP-1/GIP potentiation; incretin-based glucose-dependent insulin release |
0.5–0.8% |
Weight neutral; low hypoglycaemia risk; renal-dose adjustable |
Rare pancreatitis; heart failure risk (saxagliptin) |
|
GLP-1 Receptor Agonists |
GLP-1R agonism; insulin secretion; glucagon suppression; gastric emptying delay |
1.0–2.0% |
Weight loss 3–5 kg; CV mortality reduction; GLP-1RA class benefit |
GI side effects; injectable (most); pancreatitis risk; cost |
|
SGLT2 Inhibitors |
Renal glucose reabsorption blockade; glucosuria induction |
0.6–1.0% |
Weight loss; BP reduction; HF hospitalisation ↓; CKD protection |
Genital mycotic infections; DKA (rare); volume depletion |
|
Insulin (Basal/Bolus) |
Direct insulin receptor signalling; universal glucose metabolism regulator |
Variable (≥2.0%) |
Most potent; suitable for any stage; no dose ceiling |
Hypoglycaemia; weight gain; injection burden; patient adherence |
|
Thiazolidinediones (TZDs) |
PPARγ agonism; insulin sensitisation in adipose/muscle |
0.5–1.4% |
Durable glycaemic control; NASH benefit; HDL improvement |
Weight gain; fluid retention; fracture risk; bladder cancer (pioglitazone) |
|
Alpha-Glucosidase Inhibitors |
Intestinal carbohydrate absorption inhibition |
0.5–0.8% |
Post-prandial glucose control; weight neutral; no hypoglycaemia |
Flatulence; GI discomfort; multiple daily dosing; limited efficacy |
DPP-4: dipeptidyl peptidase-4; GLP-1RA: glucagon-like peptide-1 receptor agonist; SGLT2i: sodium-glucose cotransporter-2 inhibitor; CV: cardiovascular; GI: gastrointestinal; TZD: thiazolidinedione; HbA1c: glycated haemoglobin; BP: blood pressure; DKA: diabetic ketoacidosis.
EVIDENCE-BASED LIFESTYLE MODULATION
Dietary Interventions: Molecular Mechanisms and Clinical Evidence
Dietary patterns exert profound pharmacodynamic-like effects on the metabolic network, acting through mechanisms spanning epigenetic modulation, gut microbiome reprogramming, nuclear receptor activation, and post-translational modification of key metabolic enzymes [48]. The Mediterranean dietary pattern characterised by abundant vegetables, legumes, whole grains, fish, and monounsaturated fats from olive oil, with moderate wine and dairy reduces T2DM incidence and improves glycaemic control through multiple intersecting mechanisms.
Polyphenols abundant in the Mediterranean diet resveratrol (grapes), hydroxytyrosol and oleocanthal (olive oil), quercetin (onions, apples), and epigallocatechin gallate (green tea) activate SIRT1 deacetylase, which deacetylates and activates AMPK and PGC-1α, promoting mitochondrial biogenesis and fatty acid oxidation [49]. Oleic acid the primary MUFA in olive oil binds and activates GPR40/FFAR1 on pancreatic beta-cells, augmenting glucose-stimulated insulin secretion, while its anti-inflammatory properties via PPAR-α activation reduce NF-κB-driven cytokine production in adipose tissue.
Low-carbohydrate diets (LCD, <130g carbohydrate/day) and very-low-carbohydrate ketogenic diets (VLCKD, <50g/day) have demonstrated remarkable efficacy in T2DM management through carbohydrate-restricted glucose supply and induction of nutritional ketosis. The Virta Health prospective cohort (n=349, 2-year follow-up) demonstrated that continuous carbohydrate restriction achieved HbA1c reduction from 7.6% to 6.3%, 54% cessation of insulin therapy, and 94% reduction or elimination of sulfonylurea use with 67% achieving HbA1c <6.5% [50]. Mechanistically, ketone bodies particularly β-hydroxybutyrate function as signalling molecules beyond their fuel role: inhibiting NLRP3 inflammasome assembly (reducing IL-1β and IL-18 production), activating GPR109A/HCAR2 on immune cells, and inhibiting HDAC enzymes to promote anti-inflammatory gene expression [51].
Dietary fibre both soluble (oats, psyllium, legumes) and insoluble (whole grain cereals) — exerts glycaemic benefit through multiple mechanisms: viscous soluble fibre slows carbohydrate digestion and glucose absorption; fermentable fibres (FOS, inulin) selectively promote Bifidobacterium and Faecalibacterium prausnitzii growth, increasing SCFA production and enhancing GLP-1 and PYY secretion; insoluble fibre accelerates colonic transit, reducing post-prandial glucose excursions. A dose-response meta-analysis of 45 prospective cohorts (n=1.9 million) demonstrated 19% T2DM risk reduction per 10g/day dietary fibre increment [52].
Intermittent fasting (IF) protocols including time-restricted eating (TRE, 8–10 hour eating window), 5:2 fasting (2 days of 500 kcal restriction/week), and alternate day fasting improve insulin sensitivity through circadian alignment of feeding with peripheral tissue insulin sensitivity, promotion of autophagy via mTORC1 inhibition, and reduction in visceral adiposity. A 12-month RCT comparing TRE to standard dietary advice in T2DM (n=120) demonstrated comparable HbA1c reduction (−0.91% vs −0.97%) with superior adherence in the TRE group [53].
Physical Activity: Molecular Exercise Pharmacology
Exercise represents a multi-target intervention with pharmacological specificity at the molecular level, functioning as an activator of networks that pharmacological agents can only partially replicate [54]. Aerobic exercise activates AMPK in skeletal muscle through AMP/ATP ratio elevation, triggering GLUT4 vesicle translocation to the sarcolemma via a TBC1D4 (AS160) phosphorylation mechanism an insulin-independent glucose uptake pathway of critical importance in insulin-resistant states where the IRS-PI3K-AKT axis is impaired [55].
Resistance exercise exerts complementary metabolic effects by increasing skeletal muscle mass the primary repository for post-prandial glucose disposal through mTORC1-S6K1 signalling activation and muscle protein synthesis. The DARE trial (Resistance Exercise in Already Active Individuals with Type 2 Diabetes, n=251) demonstrated that combined aerobic and resistance training reduced HbA1c by 0.97% — significantly greater than either modality alone (aerobic: 0.51%, resistance: 0.38%) establishing the superiority of multimodal exercise prescriptions [56].
Exercise-induced myokine secretion constitutes an additional pharmacological dimension. Irisin cleaved from membrane protein FNDC5 by PGC-1α-driven gene expression during exercise — promotes adipose browning, improves insulin sensitivity, and exerts neuroprotective effects. IL-6 secreted by contracting myocytes in amounts 100-fold greater than resting acts as an exercise-induced myokine (distinct from inflammatory IL-6 from adipose/macrophages) activating AMPK-mediated glucose uptake and stimulating GLP-1 secretion from intestinal L-cells and pancreatic alpha-cells [57]. Meteorin-like (Metrnl), myonectin, and musclin represent additional exercise-responsive myokines with emerging roles in inter-organ metabolic crosstalk.
High-intensity interval training (HIIT) alternating brief periods of near-maximal effort (85–95% VO₂max, 1–4 minutes) with active recovery achieves comparable or superior metabolic outcomes to continuous moderate-intensity exercise (CME) in substantially less time, addressing a primary barrier to exercise adherence: time constraint. A meta-analysis of 50 RCTs (n=2,316 T2DM patients) demonstrated HIIT superiority over CME for HbA1c reduction (−0.19% additional reduction) and VO₂max improvement (+1.1 mL/kg/min), with comparable safety profiles [58]. The molecular basis for HIIT's metabolic efficiency involves greater AMPK activation amplitude, superior mitochondrial biogenesis stimulus via PGC-1α, and enhanced post-exercise insulin sensitivity duration (10–72 hours).
Psychosocial Interventions and Behavioural Pharmacology
Psychological distress — depression, diabetes distress, and health anxiety — affects 15–25% of individuals with T2DM and bidirectionally worsens glycaemic control through cortisol-mediated gluconeogenesis, sympathetic nervous system-driven lipolysis, and impaired medication adherence [59]. Mindfulness-based stress reduction (MBSR), cognitive-behavioural therapy (CBT), and acceptance and commitment therapy (ACT) have demonstrated HbA1c reductions of 0.2–0.5% in RCTs, mediated through cortisol attenuation, improved self-management behaviour, and enhanced executive function supporting dietary decision-making [60].
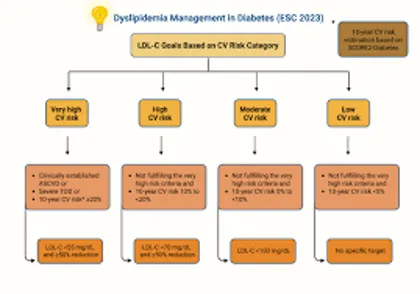
FIG.2 CURRENT TREATMENT OF DIABETES
Sleep disturbance prevalent in 60% of T2DM patients impairs insulin sensitivity by 25–40% through elevated evening cortisol, growth hormone pulsatility disruption, and increased circulating free fatty acids. A single night of sleep restriction to 4 hours produces insulin sensitivity impairment equivalent to 3 days of high-fat feeding [61]. Treatment of obstructive sleep apnoea (OSA) with continuous positive airway pressure (CPAP) reduces HbA1c by 0.3–0.6% and significantly improves nocturnal glucose profiles in CGM analyses, representing a pharmacologically distinct glycaemic intervention targeting the sleep-cortisol-metabolic axis.
Table 2: Evidence Base for Lifestyle Interventions in Type 2 Diabetes Management
|
Intervention |
Trial Evidence |
HbA1c Effect |
Metabolic Outcomes |
Implementation Notes |
|
Mediterranean Diet |
PREDIMED (n=7447); Lyon Diet Heart Study |
↓0.5–1.0% |
↓CV events 30%; ↓LDL; ↓inflammatory markers; ↓T2DM incidence 52% |
High adherence; culturally flexible; olive oil-centred; varied food patterns |
|
Low-Carbohydrate Diet (<130g/day) |
Virta Health RCT; Westman et al. 2008 |
↓1.0–1.5% |
↓TG 20–30%; ↑HDL; weight loss 5–10 kg; reduced insulin requirements |
Monitoring required for hypoglycaemia; gradual transition; micronutrient supplementation |
|
Aerobic Exercise (≥150 min/week) |
Look AHEAD trial; Umpierre et al. meta-analysis 2011 |
↓0.6–0.8% |
↓TG; ↑HDL; ↓BP 5 mmHg; improved insulin sensitivity; cardiorespiratory fitness |
Moderate intensity preferred; pre-exercise foot/CV assessment; gradual progression |
|
Resistance Training |
Castaneda et al. 2002; Sigal et al. 2007 (DARE trial) |
↓0.3–0.5% |
↑Lean mass; ↓visceral adiposity; improved glucose transporter expression (GLUT4) |
2–3 sessions/week; large muscle groups; supervision initially; avoid Valsalva |
|
Combined Aerobic + Resistance |
DARE trial (Sigal et al. 2007) |
↓0.97% |
Superior to either alone; optimal body composition; cardiovascular fitness |
Sequential same-day or alternate days; total 210–300 min/week target |
|
Intensive Lifestyle Program |
DPP (Diabetes Prevention Program) 58% risk reduction |
↓1.0–2.0% |
Weight loss ≥5% body weight; ↓T2DM incidence; ↓metabolic syndrome components |
Structured support; caloric deficit 500–750 kcal/day; behavioural counselling |
|
Sleep Optimisation (7–9 h) |
Spiegel et al.; MESA Sleep Study |
↓0.2–0.4% |
↓insulin resistance; ↓cortisol; improved leptin/ghrelin balance; ↓HbA1c |
Sleep hygiene counselling; screen apnoea; avoid late eating; consistent schedule |
|
Stress Reduction (MBSR) |
van Son et al. 2013 RCT; Hartmann et al. |
↓0.2–0.4% |
↓cortisol 14%; ↓sympathetic activation; improved medication adherence |
8-week programme; mindfulness-based; suitable adjunct to pharmacotherapy |
PREDIMED: Prevención con Dieta Mediterránea; DPP: Diabetes Prevention Program; DARE: Diabetes Aerobic and Resistance Exercise; MBSR: Mindfulness-Based Stress Reduction; TRE: Time-Restricted Eating; IOM: Institute of Medicine; OGTT: Oral Glucose Tolerance Test; PHQ-9: Patient Health Questionnaire-9.
NETWORK PHARMACOLOGY AND SYSTEMS-LEVEL DRUG INTERACTIONS
Multi-Target Drug Network Analysis
Network pharmacology applies graph theory to drug-target-disease interactions, enabling the systematic identification of synergistic drug combinations, off-target toxicity prediction, and rational polypharmacy design [62]. In the context of T2DM, construction of a disease module network mapping diabetes-associated genes (identified through GWAS loci including TCF7L2, SLC30A8, PPARG, KCNJ11, and HNF1A) onto the human protein interactome reveals that antidiabetic drugs cluster within topologically central, high-connectivity nodes (hubs) of the metabolic network, explaining both their broad efficacy and their propensity for off-target effects.
The network proximity of GLP-1R and SGLT2 to cardiovascular disease modules provides mechanistic explanation for the cardiorenal benefits observed in CVOTs, while the topological distance of DPP-4 from cardiac modules contextualises the absence of CV mortality benefit in DPP-4i trials [63]. Multi-target network analysis of metformin identifies 48 experimentally validated protein targets beyond AMPK and complex I, including HMGB1, mTOR, EGFR, and several oncogenic kinases — providing mechanistic substrate for the epidemiologically observed cancer risk reduction (25–37% reduction in colorectal, pancreatic, and hepatocellular cancers) associated with metformin use [64].
Polypharmacy analysis using network approaches has identified clinically significant drug-drug interactions (DDIs) in diabetic patients who carry on average 5–8 concurrent medications. The combination of SGLT2i with loop diuretics amplifies volume depletion risk and requires dose reduction; GLP-1RA with DPP-4i achieves no additional glycaemic benefit (as GLP-1RA already supersaturates GLP-1R, making endogenous GLP-1 preservation by DPP-4i redundant) and should generally be avoided; thiazolidinediones combined with insulin significantly increase fluid retention and heart failure risk [65]. Network pharmacology tools such as DrugBank, STRING, and DIAMOnD algorithms provide computational scaffolding for predicting and navigating these interactions.
Pharmacogenomics in T2DM: Precision Drug Selection
Pharmacogenomic variation contributes substantially to the 30–50% inter-individual variability in antidiabetic drug response and the observed differential safety profiles across ethnic groups. CYP2C9 polymorphisms particularly CYP2C9*2 and *3 alleles, with prevalence of 5–10% in European populations reduce sulfonylurea (glipizide, tolbutamide) clearance by 50–70%, dramatically increasing hypoglycaemia risk in carriers [66]. TCF7L2 rs7903146 T-allele the strongest common genetic determinant of T2DM risk predicts superior response to sulfonylureas compared to metformin, while ABCA1 rs2020927 is associated with attenuated metformin response.
The MODY genetic landscape exemplifies the clinical utility of precision pharmacogenomics. MODY3 (HNF1A mutation, ~35% of MODY) is characterised by exquisite sulfonylurea sensitivity 5-fold greater HbA1c reduction compared to metformin permitting safe and durable glycaemic control without insulin in most patients for decades after diagnosis, provided renal tubular dysfunction (Fanconi syndrome) is excluded [9]. Conversely, MODY2 (GCK mutation, ~15%) represents a benign form with mildly elevated fasting glucose that requires no pharmacotherapy (except in pregnancy), as treated glucose levels remain identical to untreated — a paradigmatic example where genetic diagnosis prevents lifelong unnecessary treatment [67].
Emerging pharmacogenomic research has begun to characterise response determinants for GLP-1RAs and SGLT2i. GLP1R polymorphisms rs10305492 (A316T), rs3765467 (G168S) modulate GLP-1RA-induced insulin secretion and weight loss response [68]. SLCO1B1 variants (impaired hepatic uptake transporter) are associated with metformin-related lactic acidosis risk in rare cases. SLC22A2 (OCT2) polymorphisms affect renal tubular metformin secretion and may modulate SGLT2i pharmacokinetics. The incorporation of pharmacogenomic panels into clinical T2DM management remains aspirational but is increasingly feasible with point-of-care genotyping costs declining below USD 100/panel.
Gut Microbiome-Drug Interactions
The gut microbiome represents an underappreciated pharmacological modulator that influences antidiabetic drug efficacy, safety, and mechanism of action. Metformin whose gastrointestinal availability far exceeds systemic concentrations after oral dosing — exerts direct antimicrobial effects that reshape the gut microbiota, increasing Akkermansia muciniphila, Bifidobacterium, and Lactobacillus and decreasing Bilophila wadsworthia [24]. Transplantation of metformin-associated microbiota into germ-free mice partially recapitulates metformin's glycaemic effects, establishing the microbiome as a pharmacodynamic mediator rather than merely a bystander.
SGLT2 inhibitors increase glycosuria, changing the luminal nutrient environment and selectively enriching glucose-consuming bacteria in the colon. Canagliflozin additionally inhibits intestinal SGLT1, elevating colonic glucose and promoting fermentative microbial activity [69]. GLP-1RAs alter gut motility, bile acid composition, and mucosal permeability in ways that secondarily modulate microbiome composition, potentially creating a virtuous cycle in which GLP-1RA-induced Akkermansia enrichment further enhances GLP-1 secretion from L-cells.
Faecal microbiota transplantation (FMT) from lean, insulin-sensitive donors to obese T2DM recipients produces transient improvements in insulin sensitivity (approximately 4–8 weeks) in controlled trials, mediated by restoration of SCFA-producing bacteria and reduction in pro-inflammatory species [70]. While FMT is not yet an approved therapeutic for T2DM, it establishes proof-of-concept for microbiome-targeted interventions as a pharmacological adjunct and motivates the investigation of next-generation synbiotics (probiotic-prebiotic combinations) targeting specific metabolic pathways.
DIGITAL HEALTH TECHNOLOGIES AND PERSONALISED MONITORING
Continuous Glucose Monitoring: Beyond HbA1c
Continuous glucose monitoring (CGM) has fundamentally transformed diabetes management by shifting the glycaemic assessment paradigm from the retrospective average (HbA1c) to dynamic, real-time glucose profiles encompassing not only mean glucose but glycaemic variability, time-in-range (TIR, 70–180 mg/dL), time-below-range (TBR, <70 mg/dL), and time-above-range (TAR, >180 mg/dL) [71]. The International Consensus on Time in Range (2019) established TIR targets of >70% for T1DM and T2DM on insulin and >50% for older/high-risk individuals metrics that correlate with HbA1c but capture pathophysiologically distinct aspects of glycaemic biology.
Retrospective CGM analysis reveals that HbA1c can mask dangerous glycaemic variability: patients with identical HbA1c values of 7.5% may exhibit TIR of 30% vs. 85% vastly different risk profiles for hypoglycaemia-related arrhythmias, nocturnal events, and impaired cognition. Coefficient of variation (CV) >36% identifies patients at clinically significant glycaemic instability regardless of HbA1c, triggering regimen reassessment [72]. Real-time CGM (rtCGM) systems — Dexcom G7, Abbott FreeStyle Libre 3 combined with insulin pump closed-loop systems (hybrid closed-loop, "artificial pancreas" technology) represent the current apex of pharmacological precision in insulin delivery.
Artificial Intelligence and Decision Support in Diabetes Management
Artificial intelligence (AI) and machine learning (ML) applications in diabetes management have progressed from predictive modelling of hypoglycaemia risk to fully autonomous insulin dosing algorithms in closed-loop systems [73]. The iLet bionic pancreas — using a Bayesian-based ML algorithm adapting insulin and glucagon delivery to individual physiology without carbohydrate counting achieved superior TIR compared to standard care in the pivotal BETA-3 trial (HbA1c 7.3% vs 7.7%), demonstrating the clinical readiness of autonomous AI-driven glycaemic control [74].
Beyond insulin delivery, ML algorithms applied to EHR data, genomic profiles, CGM time-series, and wearable sensor data can stratify T2DM patients into pharmacological responder subgroups — predicting GLP-1RA versus SGLT2i vs metformin superiority in individual patients with AUC of 0.71–0.82 in retrospective analyses [75]. Natural language processing (NLP) applied to clinical notes identifies dietary patterns, medication adherence signals, and psychosocial barriers with accuracy approaching structured questionnaires. Federated learning approaches training models across distributed health system data without centralised data sharing enable algorithm development respecting patient privacy while utilising population-scale data.
PERSONALISED TREATMENT ALGORITHM: A SYSTEMS PHARMACOLOGY FRAMEWORK
Framework Principles
The personalised treatment algorithm presented herein departs from the traditional stepwise, HbA1c-threshold-driven approach in favour of a phenotype-first, comorbidity-stratified framework grounded in four core systems pharmacology principles: (1) Target the dominant pathophysiological mechanism recognise that T2DM is pharmacologically heterogeneous and that mechanism-matched drug selection improves outcomes; (2) Integrate cardiorenal risk as a primary treatment driver select medications with proven organ-protective effects before considering glycaemic efficacy alone; (3) Incorporate lifestyle as a pharmacological synergist quantify lifestyle intervention effects in pharmacodynamic terms and integrate them into dose optimisation decisions; (4) Adapt to patient-level parameters weight, renal function, cardiac status, hypoglycaemia risk, medication burden, and patient preferences constitute pharmacokinetic-pharmacodynamic modifiers of equal importance to drug mechanisms.
The algorithm employs a hierarchical decision matrix that first assesses cardiovascular disease burden (established ASCVD, heart failure, or CKD), then metabolic parameters (HbA1c, BMI, fasting glucose), then tolerability and safety constraints, before arriving at a recommended regimen. Importantly, the algorithm explicitly incorporates lifestyle prescription as a co-equal therapeutic domain specifying dietary pattern, exercise type and volume, sleep hygiene, and psychological support rather than treating lifestyle modification as a generic recommendation preceding pharmacotherapy [76].
Cardiorenal-Risk Stratified Pathway
For patients with established atherosclerotic cardiovascular disease (ASCVD), the 2023 ADA Standards of Care, 2023 ESC Guidelines on Diabetes, Pre-Diabetes, and Cardiovascular Diseases, and 2022 AHA/ACC Guideline for T2DM and Cardiovascular Disease converge on a recommendation for GLP-1RA or SGLT2i (with proven CV benefit) independent of HbA1c or metformin use [77]. This represents a fundamental paradigm shift from glucose-centred to cardiovascular risk-centred prescribing that systems pharmacology analysis fully endorses. The mechanistic evidence is compelling: empagliflozin, liraglutide, and semaglutide reduce the primary MACE endpoint through complementary mechanisms (haemodynamic vs. anti-atherosclerotic vs. plaque-stabilising) that may be additive in combination.
For patients with heart failure (HF), SGLT2 inhibitors form the pharmacological cornerstone irrespective of ejection fraction. EMPEROR-Reduced (empagliflozin) and DAPA-HF (dapagliflozin) demonstrated 25% and 26% relative risk reductions in the composite of worsening heart failure or cardiovascular death, respectively, in HFrEF populations with and without T2DM [35]. Crucially, thiazolidinediones are absolutely contraindicated in HF due to sodium-potassium ATPase-mediated sodium and water retention; saxagliptin requires cautious use given SAVOR-TIMI hospitalisation data; and insulin doses should be conservative to minimise hypoglycaemia-triggered arrhythmias.
In CKD (eGFR 15–59 mL/min/1.73m²), SGLT2 inhibitors (particularly empagliflozin and dapagliflozin to eGFR ≥20) and finerenone provide independently additive reno-protective effects, with combination use supported by the FLOW trial (semaglutide in CKD) demonstrating 24% reduction in kidney failure [78]. Metformin can be continued to eGFR ≥30 with dose adjustment; below eGFR 30, renally-cleared agents require avoidance or reduction linagliptin (DPP-4i with exclusively biliary excretion) emerges as a uniquely kidney-safe option requiring no dose adjustment across all CKD stages.
Glycaemia-Phenotype Stratified Pathway
In patients without established CVD/CKD/HF, personalised drug selection proceeds on the basis of T2DM phenotypic cluster, HbA1c height, and body weight. Cluster 1 (SAID — severe autoimmune insulin-deficient) patients require insulin from diagnosis; Cluster 2 (SIDD — severe insulin-deficient, younger, high HbA1c) benefits from early insulin intensification or GLP-1RA with rapid titration; Cluster 3 (SIRD — severe insulin-resistant, high BMI, high HOMA-IR) responds best to insulin sensitisers (metformin, TZDs), GLP-1RAs, and SGLT2i with aggressive weight reduction lifestyle programs; Clusters 4 and 5 (mild obesity- and age-related diabetes) may achieve adequate control with metformin monotherapy combined with lifestyle intervention [7].
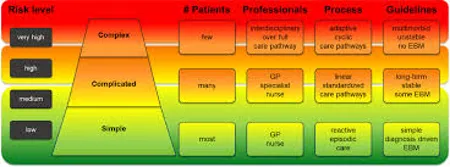
FIG.3 PERSONALISED TREATMENT OF DIABETE
For newly diagnosed T2DM with HbA1c 7.0–9.0%, metformin combined with comprehensive lifestyle intervention remains the evidence-based foundation. The GRADE study (n=5,047) — the largest comparative effectiveness RCT of second-line agents added to metformin — found that over 5 years, all four agents (insulin glargine, liraglutide, sitagliptin, glimepiride) achieved comparable initial glycaemic control, but glimepiride had higher hypoglycaemia rates, sitagliptin had higher treatment failure (HbA1c >7%), and liraglutide had highest GI side effects, while insulin and liraglutide showed superior durability informing individualised second-line selection based on priority outcomes [79].
When HbA1c exceeds 9.0% at presentation with symptomatic hyperglycaemia, dual or even triple therapy initiation outperforms stepwise addition in terms of time-to-target and beta-cell preservation a concept supported by the VERIFY trial demonstrating superior durability of upfront vildagliptin/metformin combination versus metformin monotherapy [80]. Early combination pharmacotherapy exploiting pathway complementarity metformin (hepatic glucose output) + GLP-1RA (incretin-pancreatic-central) + SGLT2i (renal glycosuria) targets three distinct nodes of the hyperglycaemia network simultaneously, reducing the probability of escape through compensatory upregulation of any single pathway.
Table 3: Personalised Treatment Algorithm Stratified by Patient Profile and Comorbidity Burden
|
Patient Profile |
First-Line Therapy |
Add-On Priority |
Lifestyle Focus |
Monitoring Parameters |
|
Newly Diagnosed, Overweight (BMI ≥27), No CV Disease |
Metformin + GLP-1RA (oral or injectable) |
SGLT2i if HbA1c ≥8.5% at 3 months; consider DPP-4i if GLP-1RA not tolerated |
Caloric restriction 500–750 kcal deficit; ≥150 min/week aerobic exercise; Mediterranean diet counselling |
HbA1c q3mo; weight monthly; lipids annually; renal function q6mo |
|
Established ASCVD + T2DM |
Metformin + GLP-1RA (proven CV benefit: liraglutide, semaglutide, dulaglutide) |
SGLT2i (empagliflozin/canagliflozin) as second agent; statin therapy |
Cardiac rehabilitation if eligible; Mediterranean diet; sodium restriction; smoking cessation |
HbA1c q3mo; BP each visit; ECG annually; eGFR & urine ACR q6mo; lipids q3–6mo |
|
Heart Failure (HFrEF or HFpEF) + T2DM |
Metformin (if eGFR ≥30) + SGLT2i (empagliflozin/dapagliflozin – HF mortality benefit) |
Avoid TZDs, saxagliptin; GLP-1RA if obese without contraindication |
Sodium restriction (<2g/day); fluid management; gentle aerobic exercise (HF-ACTION protocol); weight monitoring |
Daily weight; BNP/NT-proBNP; HbA1c q3mo; eGFR monthly initially; electrolytes |
|
Chronic Kidney Disease (eGFR 30–59 mL/min/1.73m²) |
Metformin (eGFR ≥30) + SGLT2i (finerenone for diabetic kidney disease) or GLP-1RA |
Adjust/avoid renally-cleared drugs; avoid metformin if eGFR <30; DPP-4i (linagliptin – renal-dose-free) |
Protein restriction 0.8g/kg/day; low-potassium diet if hyperkalaemia; BP <130/80 mmHg |
eGFR q3mo; urine ACR monthly; potassium; phosphate; BP each visit; HbA1c q3–6mo |
|
Elderly (≥75 years), Frail, Hypoglycaemia Risk |
DPP-4i or GLP-1RA (low hypoglycaemia risk); SGLT2i at lower dose if tolerated |
Avoid sulfonylureas; basal insulin (low dose) only if needed; simplify regimen |
Protein-sufficient diet (≥1.2g/kg/day); balance/resistance exercise (falls prevention); social engagement |
HbA1c target ≤8.0% (less stringent); cognitive assessment; fall risk; ADL function; BP (avoid hypotension) |
|
T2DM + Depression / Poor Adherence |
Simplify regimen (once-daily agents); GLP-1RA (once-weekly) preferred |
Collaborative care model; ensure stable mental health before escalating pharmacotherapy |
Motivational interviewing; digital support tools; structured group education; peer support programmes |
HbA1c q3–6mo; PHQ-9 depression screen; medication possession ratio; SMBG review |
|
Gestational Diabetes Mellitus (GDM) |
Medical nutrition therapy first; insulin (preferred pharmacotherapy) if targets unmet |
Metformin as second-line (not FDA-approved but widely used); glibenclamide third-line |
Carbohydrate-controlled diet (40–50% calories); postprandial walking; weight gain within IOM guidelines |
Fasting & 1h postprandial glucose daily; fetal growth monitoring; BP weekly; postnatal OGTT at 6–12 weeks |
ASCVD: atherosclerotic cardiovascular disease; HFrEF: heart failure with reduced ejection fraction; HFpEF: heart failure with preserved ejection fraction; CKD: chronic kidney disease; eGFR: estimated glomerular filtration rate; ACR: albumin-to-creatinine ratio; GDM: gestational diabetes mellitus; IOM: Institute of Medicine; SMBG: self-monitored blood glucose; PHQ-9: Patient Health Questionnaire-9; ADL: activities of daily living.
SPECIAL POPULATIONS IN DIABETES PHARMACOTHERAPY
Elderly Patients: Frailty, Polypharmacy, and De-intensification
Diabetes management in older adults (≥75 years) demands recalibration of the glycaemic target-treatment intensity equation to prioritise hypoglycaemia prevention, quality of life, and functional independence over tight HbA1c control [81]. Observational data from the ACCORD trial extension demonstrated a J-curve relationship between HbA1c and mortality in elderly T2DM patients, with both high (>9.0%) and low (<6.5%) HbA1c values associated with excess mortality — the latter driven by hypoglycaemia-related falls, cardiac arrhythmias, and impaired cognition. Current ADA guidelines recommend HbA1c targets of 7.5–8.0% for older adults with complex medical needs and 8.0–8.5% for those with very complex or poor health status.
Pharmacological de-intensification reducing medication burden in elderly patients who have achieved targets below recommended levels or who develop frailty is an underutilised intervention associated with improved patient wellbeing, reduced adverse effects, and lower healthcare costs. The DEPRESCRIBING framework (Determine goals; Evaluate eligibility; Pharmacological alternatives; Review risks; Engage patient; Simplify regimen; Communicate; Review; Institute safety monitoring; Broaden to non-pharmacological; Engage multidisciplinary; Gain consent) provides a structured approach applicable to geriatric diabetes management [82].
Diabetes in Pregnancy: Gestational and Pregestational
Pharmacological management of diabetes in pregnancy operates under strict constraints imposed by teratogenicity concerns, altered pharmacokinetics of pregnancy (increased renal clearance, expanded volume of distribution, changed protein binding), and fetal glucose-insulin dynamics [83]. Insulin remains the gold-standard pharmacotherapy across all trimesters with human insulin analogues (aspart, lispro, detemir) demonstrating reassuring safety profiles in RCTs. Metformin, while not FDA-approved for GDM, is used as second-line therapy in many international guidelines and demonstrates non-inferiority to insulin for GDM glycaemic control (MiG trial) with important caveats: placental transfer exposes the fetus to metformin concentrations equal to maternal plasma, and long-term follow-up data suggest potential effects on offspring insulin resistance and adiposity.
SGLT2 inhibitors are contraindicated throughout pregnancy due to fetal renal tubular SGLT2 expression (particularly in the second and third trimesters) — glycosuria promotes fetal diuresis and potential oligohydramnios. GLP-1RAs lack adequate human pregnancy safety data and are not recommended. Thiazolidinediones are contraindicated. This pharmacological scarcity in pregnancy reinforces the paramount importance of lifestyle intervention structured medical nutrition therapy achieving individualised carbohydrate targets, gestational weight gain within IOM guidelines, and post-prandial walking as the primary therapeutic modality in GDM.
Type 1 Diabetes: Adjunct Therapy and Technology Integration
While insulin replacement therapy remains the cornerstone of T1DM management, adjunct non-insulin pharmacotherapy offers incremental glycaemic benefits in selected patients. SGLT2 inhibitors (empagliflozin, dapagliflozin the latter FDA-approved as T1DM adjunct until label revision) reduce HbA1c by 0.4–0.6% and body weight by 2–3 kg in T1DM, but carry a 3–6-fold increased risk of diabetic ketoacidosis (DKA) — necessitating patient education regarding sick-day rules, ketone monitoring, and insulin dose maintenance [84]. This risk was deemed sufficient for European Medicines Agency label revision restricting T1DM use for selected patients only, underscoring the critical importance of system-level safety pharmacovigilance.
Pramlintide a synthetic amylin analogue reduces post-prandial glucose excursions in T1DM by complementing insulin through gastric emptying delay, post-prandial glucagon suppression, and central appetite reduction. Hybrid closed-loop systems integrating CGM, insulin pump, and control algorithms (MiniMed 780G, Control-IQ, Omnipod 5) represent the frontier of T1DM management, achieving TIR of 70–80% compared to 50–65% with conventional insulin pump therapy, with breakthrough improvements in nocturnal hypoglycaemia and sleep quality [85].
PHARMACOVIGILANCE, SAFETY MONITORING, AND DRUG INTERACTIONS
The complex pharmacological landscape of diabetes management characterised by multi-drug regimens spanning antidiabetic agents, cardiovascular medications, renally-active drugs, and emerging targeted therapies mandates a systematic pharmacovigilance framework that extends beyond regulatory adverse event reporting to encompass proactive drug-drug interaction screening, population-level safety signal detection, and individual patient risk stratification [86].
Real-world pharmacovigilance data from national registries have yielded important safety signals complementing RCT-derived evidence. The disproportionality analysis from FAERS (FDA Adverse Event Reporting System) identified a 2.3-fold elevated reporting odds ratio for fournier's gangrene (necrotising fasciitis of the genitoperineum) with SGLT2i use — prompting FDA label updates in 2018 and clinical guidance for prompt evaluation of genital infections in SGLT2i-treated patients [87]. Similarly, GLP-1RA-associated pancreatitis signals from FAERS, while not substantiated in large CVOTs, necessitate clinical vigilance particularly in patients with prior pancreatitis, gallstones, or hypertriglyceridaemia.
Drug-drug interaction management in diabetic polypharmacy requires systematic screening. Clinically significant interactions include: (1) Fluoroquinolones (ciprofloxacin, levofloxacin) causing both hypoglycaemia and hyperglycaemia through dual KATP channel closure and pancreatic beta-cell toxicity; (2) Beta-blockers masking sympathoadrenergic hypoglycaemia warning symptoms (tachycardia, tremor) while preserving sweating, necessitating patient education particularly in insulin-treated patients; (3) Corticosteroids dramatically increasing insulin requirements by 30–100% through transient post-prandial hyperglycaemia requiring insulin regimen adjustment; (4) Thiazide diuretics at high doses impairing glucose tolerance through hypokalaemia-mediated insulin secretion impairment [65].
Renal function monitoring represents a critical safety parameter across the antidiabetic pharmacopeia. Metformin requires eGFR assessment at baseline and every 6–12 months, with dose halving at eGFR 30–45 and cessation below 30. SGLT2i require baseline eGFR (minimum eGFR 20 for empagliflozin/dapagliflozin per current labels) and hold during acute illness, radiographic contrast procedures, and major surgery to prevent acute kidney injury superimposed on volume depletion. DPP-4 inhibitors (except linagliptin) require dose reduction at eGFR <45 (sitagliptin), <50 (saxagliptin), or <30 (alogliptin). Sulfonylureas with active renal metabolites (glibenclamide, glimepiride) should be avoided in significant CKD.
FUTURE DIRECTIONS IN DIABETES SYSTEMS PHARMACOLOGY
Multi-Omics Integration and Precision Endocrinology
The convergence of genomics, transcriptomics, proteomics, metabolomics, and microbiomics collectively termed multi-omics with systems pharmacology modelling promises a transformative paradigm shift toward true precision endocrinology [88]. The T2D-GENES Consortium, Human Islet Research Network (HIRN), and Type 1 Diabetes TrialNet have generated multi-omics datasets enabling the construction of dynamic network models of beta-cell dysfunction that predict disease progression trajectories and pharmacological response profiles with unparalleled granularity. Metabolomic signatures particularly branched-chain amino acid (BCAA) elevations, ceramide ratios, acylcarnitine profiles, and specific bile acid subspecies — have emerged as biomarkers of insulin resistance subtype, predicting differential GLP-1RA versus SGLT2i response with clinical utility approaching pharmacogenomic markers.
Proteomics-based cardiovascular risk stratification in T2DM leveraging high-throughput aptamer-based platforms (SomaScan, Olink) measuring thousands of circulating proteins has identified novel biomarker panels (GDF-15, troponin T, NT-proBNP, IGFBP-2, and others) that improve cardiovascular risk prediction beyond traditional Framingham-based models by 15–25%, potentially enabling earlier initiation of cardioprotective pharmacotherapy in high-risk subgroups before ASCVD manifestation [89].
RNA Therapeutics and Gene-Based Interventions
The mRNA therapeutic revolution catalysed by COVID-19 vaccine development has opened transformative possibilities for diabetes pharmacology. mRNA encoding GLP-1, insulin, or glucokinase could theoretically enable sustained, regulated protein production following lipid nanoparticle delivery to hepatocytes bypassing the injection burden of protein therapeutics while enabling precise tissue targeting and programmable expression duration [90]. Antisense oligonucleotides (ASOs) targeting ANGPTL3 (reducing triglycerides and LDL), PCSK9 (LDL reduction), and hepatic glucagon receptor (reducing fasting glucose) are in advanced clinical development as adjuncts to diabetes-associated dyslipidaemia and hyperglycaemia management.
CRISPR-Cas9 genome editing applications in T2DM research include correction of MODY-causing mutations in patient-derived iPSCs differentiated into insulin-producing beta-like cells providing a potential autologous cell replacement therapy. TCF7L2 risk variant modification in beta-cells enhances insulin secretion in vitro, while PPARG gain-of-function editing could phenocopy TZD effects without systemic side effects [91]. While clinical translation of genomic editing in diabetes remains distant, these platforms will substantially accelerate the identification and validation of causal therapeutic targets identified through systems pharmacology network analyses.
Digital Therapeutics and Behavioural Pharmacology Integration
Digital therapeutics (DTx) software-based medical interventions delivering evidence-based therapeutic interventions have achieved regulatory approval (FDA, CE mark) for T2DM management. Virta Health's continuous care model and Noom Med combine behavioural psychology, dietetic support, continuous CGM feedback, and telemedicine into integrated digital platforms demonstrating HbA1c reductions of 1.0–2.3% and medication reduction outcomes equivalent to intensive clinic-based interventions at substantially lower cost and greater accessibility [92]. BlueStar (Welldoc) the first prescription digital therapeutic for T2DM provides algorithm-driven coaching, medication reminders, and pattern recognition in CGM/SMBG data, achieving 1.9% HbA1c reduction in an FDA-reviewed pivotal study.
The pharmacology-behavioural psychology interface offers untapped synergistic potential. GLP-1RAs reduce not only appetite but food reward valuation through mesolimbic GLP-1R activation a psychopharmacological mechanism that potentiates dietary adherence and may reduce addictive behaviour [93]. This neurobiological synergy between pharmacotherapy and behavioural intervention provides a mechanistic rationale for co-prescribing GLP-1RAs with structured behavioural weight management programs an integration now codified in the 2023 ADA/EASD consensus statement. The pharmaceutical-behavioural ecosystem, integrating medication, digital feedback, and psychological support within a coherent systems model, represents the practical implementation of systems pharmacology in clinical diabetes care.
CONCLUSIONS
The management of diabetes mellitus in its heterogeneous phenotypic manifestations and complex comorbidity contexts demands a therapeutic paradigm that matches the biological complexity of the disease itself. Systems pharmacology, by integrating molecular target networks, multi-omics heterogeneity, clinical pharmacology, lifestyle biology, and digital health technologies into a coherent analytical framework, provides exactly this paradigm. The convergence of multiple mechanistic streams reviewed in this paper yields actionable conclusions.
First, the cardiorenal protective effects of GLP-1 receptor agonists and SGLT2 inhibitors established through convergent cardiovascular outcome trial evidence and mechanistic network analysis elevate these agents to the status of disease-modifying cardiovascular medicines, warranting their use as first-line or early-add-on therapy in any T2DM patient with CVD, heart failure, or CKD, independent of baseline HbA1c. Second, the molecular pharmacology of lifestyle interventions operating through AMPK, SIRT1, myokine signalling, gut microbiome reprogramming, and epigenetic modification warrants their quantitative integration into treatment planning as pharmacologically equivalent to second-line oral antidiabetic agents. Third, pharmacogenomics and T2DM phenotypic clustering enable the prospective identification of pharmacological responder subgroups, transforming the historically empirical trial-and-error approach to drug selection into a mechanism-matched, evidence-anchored precision prescribing process.
The personalised treatment algorithm presented herein stratified by cardiorenal comorbidity, metabolic phenotype, patient-level pharmacokinetic parameters, and lifestyle capability operationalises these systems pharmacology insights into a clinically deployable framework applicable across primary and specialist care settings. As precision medicine technologies polygenic risk scoring, CGM-derived phenotyping, AI-driven decision support, and multi-omics biomarker panels achieve clinical feasibility, the framework presented here provides the conceptual scaffold into which these tools can be progressively integrated.
The transformation of diabetes care from a reactive, complication-driven model to a proactive, systems-informed, precision therapeutic paradigm is not merely a scientific aspiration but a clinical imperative demanded by the scale of global diabetes burden, the preventable nature of its complications, and the rich mechanistic pharmacology available to those who approach it with sufficient intellectual breadth. Systems pharmacology offers that breadth.
REFERENCES





Edapalapati Venkata Naga Nithin, Raveendra N, Binaya Kumar Sethy, Darshana Vinod Rane, Ayesha Jamal, A Systems Pharmacology Approach to Diabetes Management: Integrating Drug Therapy, Lifestyle Modulation, and Personalised Treatment Algorithm , Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5483-5510. https://doi.org/10.5281/zenodo.20790752
 10.5281/zenodo.20790752
10.5281/zenodo.20790752