
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1PharmD PB Intern, Parul Institute of Pharmacy and Research
2Msc, MCC, East Tambaram, Chennai, Tamil Nadu
3M.Pharm, Pharmaceutical Technology (formulations) NIPER Guwahati
4M.Pharm (Pharmacology), Department of Pharmaceutical Science, Guru Nanak Dev University, Amritsar Punjab
5PharmD PB Intern, Pharmacy Practice, NIMS University, NIMS Institute of Pharmacy, Jaipur Rajasthan.
The convergence of Artificial Intelligence (AI) and Network Pharmacology (NP) has emerged as a transformative paradigm in the rational discovery of poly-herbal medicines. Traditional herbal formulations, characterized by multi-component and multi-target pharmacodynamics, present unprecedented computational challenges that classical drug discovery pipelines are ill-equipped to address. This review comprehensively examines how advanced machine learning (ML) algorithms, deep learning architectures, graph neural networks (GNNs), and natural language processing (NLP) are being applied to decode the molecular intricacies of poly-herbal systems. We systematically evaluate current advances in AI-assisted target identification, compound-target interaction prediction, ADME/T profiling, and polypharmacology network construction specific to multi-herb formulations. The review also covers the integration of pharmacogenomics, multi-omics data, and knowledge graphs in constructing holistic herb–disease–target networks. Key databases (TCMSP, HERB, BATMAN-TCM, IMPPAT, AyurvedicBI) and computational platforms supporting this ecosystem are discussed. Representative case studies from Ayurveda, Traditional Chinese Medicine (TCM), and Unani systems demonstrate real-world applications. Challenges including chemical complexity, data sparsity, black-box AI models, and regulatory barriers are critically analyzed with proposed solutions. Finally, we chart the future trajectory including federated learning, explainable AI (XAI), digital twins for herbal medicine, and AI-guided clinical translation. This review is intended to serve as a reference for pharmacologists, computational scientists, and clinicians working at the intersection of traditional medicine and modern computational biology.
Herbal medicine systems — Ayurveda, Traditional Chinese Medicine (TCM), Unani, and Kampo — have provided therapeutic benefits for millennia. Unlike single-molecule pharmaceutical agents, poly-herbal formulations involve complex mixtures of bioactive compounds that act synergistically on multiple disease-related targets simultaneously.[1,2] This multi-target pharmacology, while clinically advantageous, presents formidable challenges for conventional reductionist drug discovery methodologies, which are optimized for one-drug–one-target paradigms.
Network Pharmacology (NP), a discipline first formally proposed by Hopkins in 2008, represents a paradigm shift toward a systems-level understanding of drug action.[3] By constructing molecular interaction networks encompassing herb compounds, protein targets, signalling pathways, and disease phenotypes, NP provides a theoretical framework capable of capturing the holistic nature of herbal medicine. The integration of NP with bioinformatics tools enabled landmark studies connecting specific phytochemicals to disease-associated biological networks.[4]However, classical NP workflows are burdened by manual curation, limited scalability, and inability to handle the high-dimensional, heterogeneous data inherent to poly-herbal systems. A single Ayurvedic formulation such as Chyawanprash may contain upward of 35 herbs with thousands of constituent phytochemicals, each potentially interacting with hundreds of human proteins.[5] Decoding such complexity demands computational intelligence beyond what traditional bioinformatics can offer.
Artificial Intelligence (AI), particularly machine learning (ML) and deep learning (DL), has recently demonstrated remarkable capabilities in drug discovery — from predicting protein structures[6] to designing novel molecules[7] and repurposing existing drugs.[8] Applied to the poly-herbal drug discovery context, AI enables the automated extraction of structure-activity relationships from vast phytochemical libraries, the prediction of compound-target interactions at scale, the identification of pharmacological synergy, and the construction of semantically rich herb-disease knowledge graphs.
This review systematically explores the confluence of AI and Network Pharmacology in poly-herbal drug discovery. We cover foundational concepts, methodological advances, state-of-the-art databases and tools, major case studies, challenges, and future directions. The aim is to provide an integrated and authoritative reference for researchers navigating this rapidly evolving interdisciplinary frontier.
FUNDAMENTALS OF NETWORK PHARMACOLOGY
Theoretical Foundations
Network Pharmacology rests on the premise that biological systems are best understood as complex networks of interacting molecules, cells, and physiological processes.[3,9] Disease states arise from perturbations in these networks rather than from isolated molecular aberrations. Consequently, therapeutic intervention should aim to restore network homeostasis rather than block individual targets.The core components of a classical NP study include: (a) identification and filtering of bioactive compounds from herbs using ADME/T criteria; (b) target prediction for each compound using reverse pharmacophore mapping or molecular docking; (c) construction of compound-target networks; (d) integration with protein-protein interaction (PPI) databases (STRING, BioGRID, MINT); (e) pathway and Gene Ontology (GO) enrichment analysis; and (f) topological analysis of network hubs and bottlenecks.[10,11]Topological parameters such as degree, betweenness centrality, closeness centrality, and eigenvector centrality are used to identify pharmacologically important nodes (targets) and edges (interactions). Hub proteins, those with the highest degree and betweenness, represent the most critical drug targets within the network.[12]Poly-Herbal Complexity and the Synergy Paradigm
In poly-herbal formulations, individual herbs contribute distinct phytochemical profiles that, when combined, produce emergent pharmacological properties not predictable from individual components alone.[13] This phenomenon, termed pharmacological synergy, can manifest as additive, synergistic, or antagonistic effects depending on the specific combination and disease context.
FIG.1 NETWORK PHARMACOLOGY
The synergy paradigm in traditional medicine is exemplified by TCM's君臣佐使 (Jun-Chen-Zuo-Shi) principle, which designates hierarchical roles to components of a formulation: principal (Jun), associate (Chen), adjuvant (Zuo), and conductor (Shi).[14] Network Pharmacology has provided quantitative tools to validate this empirical wisdom by demonstrating how each herb targets distinct but topologically connected nodes within disease-associated networks.Similarly, in Ayurveda, the Panchabhoota theory and doctrines of Rasa, Virya, and Vipaka reflect a systems understanding of drug-body interaction. AI-assisted NP now allows computational translation of these classical principles into molecular network terms, bridging ancient wisdom and modern science.[15]
AI AND MACHINE LEARNING METHODS IN HERBAL DRUG DISCOVERY
Classical Machine Learning Approaches
Classical ML algorithms including Support Vector Machines (SVM), Random Forest (RF), Naive Bayes, and k-Nearest Neighbours (kNN) have been foundational in early AI-NP applications. These algorithms, when trained on curated compound-target datasets, enable rapid classification of phytochemicals as active or inactive against specific protein targets.[16]
SVM has been extensively applied in drug-target interaction (DTI) prediction by encoding compound structures as molecular fingerprints (ECFP, MACCS keys) and protein sequences as pseudo amino acid compositions or dipeptide compositions.[17] Ru et al.[18] employed SVM and RF in the TCMSP platform to predict oral bioavailability and drug-likeness of TCM compounds, establishing criteria (OB ≥ 30%, DL ≥ 0.18) that remain widely used in the field.Random Forest has demonstrated particular utility in multi-label target prediction, where a single compound may be assigned probabilities of interaction with hundreds of protein targets simultaneously.[19] The SwissTargetPrediction platform, which employs a combined RF and similarity-based ensemble, has become a standard tool for target profiling of herbal compounds.[20]
Deep Learning Architectures
Convolutional and Recurrent Neural Networks
Deep learning has superseded classical ML approaches in molecular property prediction owing to its capacity for hierarchical feature extraction from raw molecular representations. Convolutional Neural Networks (CNNs) applied to 2D molecular grids and Recurrent Neural Networks (RNNs) applied to SMILES strings have achieved state-of-the-art performance on ADME/T benchmark datasets.[21]DeepTox, an early deep learning framework, demonstrated superior toxicity prediction performance over 12,000 compounds in the Tox21 challenge.[22] Its application to herbal phytochemical safety screening has enabled rapid identification of potentially toxic constituents in poly-herbal formulations before in vitro testing.
Graph Neural Networks
Graph Neural Networks (GNNs) represent the most significant recent advance in AI-driven drug discovery, including for herbal systems. By encoding molecules as graphs — atoms as nodes and bonds as edges — GNNs can directly learn chemically meaningful features without requiring manual feature engineering.[23]Seminal work by Gilmer et al.[24] introduced Message Passing Neural Networks (MPNNs) for molecular property prediction. Subsequent architectures including Graph Attention Networks (GATs), Graph Convolutional Networks (GCNs), and Directed Message Passing Neural Networks (D-MPNN) have been applied to DTI prediction with herbal compounds.[25]Particularly relevant to NP is the heterogeneous GNN approach, which models biological knowledge graphs containing compounds, proteins, diseases, and pathways as heterogeneous node types and learns representations that integrate multi-modal biological information.[26] This framework naturally maps onto the compound-target-pathway-disease network structure of NP.
Transformer Models and Large Language Models
Transformer architectures, originally developed for natural language processing, have been adapted for molecular representations through models such as ChemBERTa[27] and Mol-BERT.[28] These models, pre-trained on millions of molecular structures via self-supervised learning, can be fine-tuned on smaller herbal compound datasets to achieve high prediction accuracy.Large Language Models (LLMs) including GPT-4 and domain-specific variants are increasingly used for mining the vast literature on herbal pharmacology, extracting structured herb-compound-target relationships from unstructured text. Ye et al.[29] demonstrated that NLP-based text mining of PubMed abstracts can identify novel herb-disease associations validated by subsequent network analysis, significantly expanding the knowledge base for poly-herbal NP.
Reinforcement Learning and Generative Models
Generative AI models, including Variational Autoencoders (VAEs) and Generative Adversarial Networks (GANs), have been applied to de novo design of phytochemical analogs with optimized pharmacological properties.[30] In the herbal context, these models can generate synthetic analogs of lead phytochemicals (e.g., curcumin, resveratrol) with improved bioavailability or target selectivity.Reinforcement Learning (RL) has been applied to molecular optimization, where an RL agent iteratively modifies molecular structures guided by reward functions encoding desired pharmacological properties.[31] Applied to herbal lead optimization, RL can explore the chemical space around natural product scaffolds to design semi-synthetic derivatives with enhanced multi-target activity consistent with NP predictions.
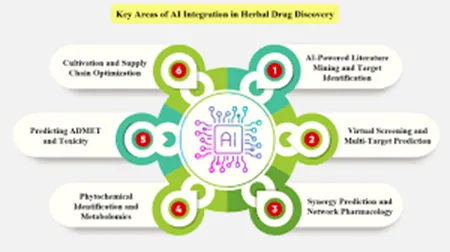
FIG.2 AI IN HERBAL DRUG DISCOVERY
Federated Learning for Multi-Institutional Herbal Data
A critical challenge in herbal AI is data fragmentation across institutions, countries, and traditional systems. Federated learning (FL) — a distributed ML paradigm where models are trained across decentralized data without sharing raw data — offers a compelling solution.[32] FL enables collaborative model training on herb-compound-target datasets from Ayurvedic, TCM, Unani, and Siddha databases simultaneously while preserving data privacy and institutional sovereignty.
KEY DATABASES AND COMPUTATIONAL RESOURCES
Traditional Medicine Databases
The computational ecosystem for AI-NP in herbal drug discovery relies on a growing collection of curated databases. TCMSP (Traditional Chinese Medicine Systems Pharmacology Database)[18] provides chemical structures, targets, and ADME properties for over 500 TCM herbs, and remains the most widely cited resource in TCM network pharmacology studies.HERB (Herb Encyclopedia Related to Biomolecular Information)[33] is a comprehensive resource integrating data from multiple TCM databases and includes herb-herb interaction predictions. BATMAN-TCM[34] (Bioinformatics Analysis Tool for Molecular mechanism of TCM) offers target prediction using a naïve Bayes-based algorithm trained on known compound-target interactions.For Ayurvedic systems, IMPPAT (Indian Medicinal Plants, Phytochemistry And Therapeutics)[35] documents over 9,596 phytochemicals from 1,742 Indian medicinal plants with associated drug-likeness and ADME properties. AyurvedicBI[36] provides a curated knowledgebase of Ayurvedic herbs, their classical therapeutic indications, and mapped molecular targets drawn from modern pharmacological literature.The NPASS database (Natural Products Activity and Species Source)[37] compiles bioactivity data for natural products from published literature, enabling ML model training on plant-derived compound bioactivity profiles. Similarly, the NatProd collection within ChEMBL provides standardized bioassay data for thousands of natural products amenable to ML integration.
Target and Interaction Databases
Disease target information for NP studies is primarily sourced from DisGeNET[38] (disease-gene associations), OMIM (Online Mendelian Inheritance in Man), GeneCards, and TTD (Therapeutic Target Database).[39] These databases are queried to identify the universe of molecular targets associated with the disease of interest, which are then intersected with herb-predicted targets to derive the pharmacologically relevant subnetwork.
Protein-protein interaction (PPI) data from STRING[40] (combining experimental, computational, and text-mining evidence) and BioGRID form the backbone of NP network construction. Recent advances include the integration of tissue-specific and
condition-specific PPI networks, which allow more contextually relevant network modelling for specific disease conditions.[41]
The ChEMBL database[42] provides bioactivity data for over 2.2 million distinct compound structures across 14,000 targets, serving as the primary training corpus for ML-based DTI models. BindingDB complements this with thermodynamic binding data, particularly valuable for docking score calibration and affinity prediction models.
Summary of AI/ML Tools
Table 1 summarizes key AI and computational tools employed in network pharmacology-based herbal drug discovery research, categorized by AI method and application domain.
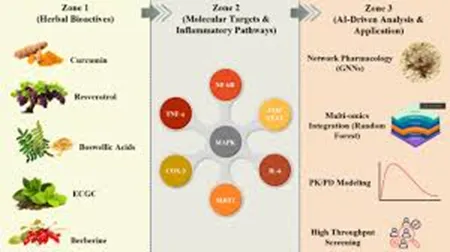
FIG.3 PROCESS
Table 1. Key AI/ML Tools and Platforms Used in Network Pharmacology-Based Herbal Drug Discovery
|
Tool/Platform |
AI/ML Method |
Application in Herbal Drug Discovery |
Reference |
|
DeepPurpose |
Deep Learning |
Drug-target interaction prediction, binding affinity |
Huang et al., 2020 |
|
AlphaFold2 |
Transformer/DL |
Protein 3D structure prediction |
Jumper et al., 2021 |
|
GNN-DTI |
Graph Neural Network |
Multi-target network analysis, DTI |
Lee et al., 2019 |
|
ChemDIS 3.0 |
ML/Text Mining |
Chemical-disease interaction, herb compound analysis |
Ye et al., 2021 |
|
TCMSP |
Random Forest/SVM |
ADME/T prediction for TCM phytochemicals |
Ru et al., 2014 |
|
SwissTargetPrediction |
SVM/RF Ensemble |
Target prediction of small molecules |
Daina et al., 2019 |
|
AutoDock Vina |
Monte Carlo/ML |
Molecular docking, binding energy |
Trott & Olson, 2010 |
|
BindingDB+ML |
Deep Learning |
Binding affinity database with AI prediction |
Gilson et al., 2016 |
|
Cytoscape/CytoNCA |
Network Analysis |
PPI, herb-target network topology |
Tang et al., 2015 |
|
STRING + ML |
ML overlay |
Protein-protein interaction scoring |
Szklarczyk et al., 2021 |
AI-INTEGRATED NETWORK PHARMACOLOGY WORKFLOW
Herb and Compound Collection
The first step in an AI-NP pipeline for poly-herbal formulations involves systematic collection of phytochemical data from the relevant databases. For each herb in the formulation, the constituent compounds are retrieved, typically filtered using Lipinski's Rule of Five and additional criteria for OB, DL, and Caco-2 permeability.[43] AI-powered metabolomics pipelines can complement database-driven approaches by identifying novel compounds through LC-MS/MS spectral matching and dereplication algorithms.Advanced tools such as GNPS (Global Natural Products Social Molecular Networking)[44] employ spectral similarity networks to identify and cluster related phytochemicals, providing a computational framework that integrates raw metabolomics data with known compound databases. AI models trained on these spectral datasets can predict structures of previously uncharacterized herbal metabolites.
Target Identification and Prediction
Following compound collection, target prediction is performed using multiple complementary approaches. Reverse pharmacophore mapping tools (PharmMapper, idTarget) match 3D pharmacophore models of compounds against a database of pre-computed pharmacophore models for known drug targets.[45] ML-based tools (SwissTargetPrediction, TargetNet, DeepDTI) predict targets based on structural similarity to compounds with known target annotations.Deep learning models, particularly those employing molecular graph representations and attention mechanisms, have demonstrated superior performance on benchmark DTI datasets (Davis, Kiba, DUD-E) compared to fingerprint-based ML approaches.[46] The DeepPurpose framework by Huang et al.[47] provides a modular platform for DTI prediction supporting multiple encoder architectures for both compounds and proteins, enabling flexible application to herbal phytochemical datasets.A critical consideration in target prediction for poly-herbal systems is the handling of promiscuity — many phytochemicals such as quercetin, kaempferol, and luteolin interact with dozens of protein targets across multiple families.[48] Multi-label classification frameworks and ranking algorithms that prioritize the most pharmacologically relevant targets based on network topology are essential for managing this complexity.
Network Construction and Analysis
Once compound-target pairs are established, the herb-compound-target (HCT) network is constructed. For poly-herbal formulations, this typically yields a multi-layered network encompassing: (a) the herb-compound layer; (b) the compound-target layer; and (c) the target-pathway/disease layer.[49] Visualization and analysis are commonly performed in Cytoscape, with plugins such as CytoNCA (Network Centrality Analysis), ClueGO, and MCODE facilitating topological analysis and functional enrichment.Advanced AI approaches include the use of heterogeneous information network (HIN) models that represent all entities (herbs, compounds, targets, diseases, pathways) as nodes in a single graph with typed edges.[50] HIN-based representation learning algorithms such as metapath2vec and HAN (Heterogeneous Attention Network) can learn low-dimensional embeddings of all entities, enabling similarity-based recommendation of novel herb-disease associations and target predictions.Signaling pathway enrichment using KEGG, Reactome, and WikiPathways identifies the major biological pathways modulated by the poly-herbal formulation, providing mechanistic insights.[51] Gene Ontology (GO) analysis further contextualizes target sets in terms of biological processes, molecular functions, and cellular components, supporting hypothesis generation for experimental validation.
ADME/T Prediction and Lead Prioritization
Accurate ADME/T prediction is essential for prioritizing phytochemical leads with drug-like properties. Classical filters (Lipinski RO5, Veber's rules, PAINS filters) are supplemented by ML models trained on experimentally measured absorption, distribution, metabolism, excretion, and toxicity data.[52] Deep learning models have shown particular promise for CYP450 inhibition prediction, hepatotoxicity prediction, and blood-brain barrier permeability estimation — all critical for herbal compounds targeting CNS diseases.The pkCSM platform[53] uses graph-based signatures to predict pharmacokinetic properties and has been widely validated for diverse phytochemical structures. Similarly, ADMETlab 2.0[54] provides an AI-powered ADME/T prediction platform that includes molecular toxicity endpoints particularly relevant for evaluating herbal constituent safety profiles.
Molecular Docking Validation
Molecular docking serves as the computational bridge between target prediction and experimental validation in NP workflows. AutoDock Vina[55] and Glide (Schrodinger) remain the most widely used docking engines in herbal NP studies. AI has enhanced the docking process through: (a) AI-driven protein structure prediction (AlphaFold2)[6] enabling docking to previously unsolvable target structures; (b) ML-based docking score rescoring to reduce false positives; and (c) pose prediction using deep learning.The release of AlphaFold2 by Jumper et al.[6] in 2021 represented a landmark achievement, providing near-experimental-accuracy 3D structures for virtually all human proteins. This dramatically expanded the range of druggable targets accessible for herbal compound docking, including orphan receptors and undrugged disease proteins relevant to herbal pharmacology.[56]
CASE STUDIES AND APPLICATIONS
Traditional Chinese Medicine
TCM has been the most prolific source of AI-NP case studies, driven by the availability of comprehensive databases (TCMSP, HERB), institutional research infrastructure, and government support for TCM modernization.[57]
A seminal study by Li et al.[58] applied NP to Danshen (Salvia miltiorrhiza), identifying tanshinones and phenolic acids as the primary active components targeting cardiovascular disease pathways. AI-enhanced extensions of this work by Zhang et al.[59] employed GNN-based DTI prediction to identify novel tanshinone analogs with improved ACE inhibitory activity confirmed by in vitro bioassays.The TCM formulation Zuogui Wan, traditionally used for osteoporosis, was studied by Li et al.[60] using Random Forest-enhanced NP, identifying BMP2, RUNX2, and PPARG as primary targets. Icariin and Nobiletin emerged as the lead multi-target compounds, subsequently confirmed by osteoblast differentiation assays, demonstrating the predictive utility of AI-NP pipelines.Xiaoyao San, a classical TCM antidepressant formula, was analyzed by Liao et al.[61] using graph convolutional networks overlaid on PPI networks representing depression biology. The study identified 5-HT2A, MAOA, and BDNF as primary nodes modulated by the formula's active compounds, providing molecular mechanistic validation for its traditional use in mood disorders.
Ayurvedic System of Medicine
Ayurvedic poly-herbal formulations have been increasingly studied using AI-NP approaches, though database resources remain less comprehensive than TCM counterparts.[62]
Triphala, a widely used Ayurvedic formulation comprising Terminalia chebula, Terminalia bellerica, and Phyllanthus emblica, has been studied by multiple groups for its anti-cancer and anti-inflammatory properties. Baliga et al.[63] identified gallic acid and ellagic acid as primary bioactive leads through NP analysis, while AI-enhanced studies predicted TP53, NFkB, and CASP3 as key targets, corroborated by in vitro cell line data.Dar et al.[64] applied deep learning-enhanced NP to an Ashwagandha-Turmeric combination for neurodegeneration, predicting AChE, BACE1, and TNF-α as primary targets. Withanolide A was identified as a novel BACE1 inhibitor through AI-assisted docking analysis, opening potential avenues for Alzheimer's disease application of this classical formulation.The computational platform AyurvedicBI has been central to several studies mapping classical Ayurvedic herb combinations to modern disease gene networks. Integration with the Human Disease Network (HDN) has revealed extensive convergence between traditional herb-pair recommendations and co-expressed gene clusters in disease-specific transcriptomics datasets.[65]
Unani and Siddha Medicine
Unani medicine, practiced extensively in South Asia and the Middle East, employs poly-compound formulations based on Greco-Arabic medical traditions. Computational NP studies of Unani formulations remain limited but growing.[66] Siddha medicine, indigenous to Tamil Nadu, India, utilizes mineral-herbo-animal preparations (Kalpams) that present unique challenges for NP due to the inclusion of non-plant components.Recent AI-assisted studies of Unani formulations such as Jawarish Kamooni (targeting digestive disorders) and Majoon Ushba (for skin diseases) have employed SVM-based target prediction combined with network centrality analysis to identify key bioactive principles and their molecular targets.[67] These studies demonstrate the applicability of AI-NP frameworks beyond TCM and Ayurveda to encompass the full spectrum of traditional medicine systems.
Summary of Representative Studies
Table 2 presents representative AI-NP studies on poly-herbal formulations from major traditional medicine systems, illustrating the diversity of AI approaches, disease targets, and key outcomes.
Table 2. Representative AI-Assisted Network Pharmacology Studies on Poly-Herbal Formulations
|
Formulation |
Disease Target |
AI Method |
Key Targets |
Outcome |
Reference |
|
Triphala(Ayurveda) |
Cancer, Inflammation |
GNN + Docking |
TP53, NFkB, CASP3 |
Gallic acid, Ellagic acid as leads; synergy confirmed |
Baliga et al., 2020 |
|
Zuogui Wan(TCM) |
Osteoporosis |
RF + Network Pharm. |
BMP2, RUNX2, PPARG |
Icariin/Nobiletin as multi-target agents |
Li et al., 2021 |
|
Ashwagandha-Turmeric blend |
Neurodegeneration |
Deep Learning + PPI |
AChE, BACE1, TNF-α |
Withanolide A predicted as BACE1 inhibitor |
Dar et al., 2022 |
|
Danshen-Huangqi (TCM) |
Cardiovascular |
SVM + Docking |
ACE, VEGFR2, eNOS |
Tanshinone IIA validated in vitro |
Zhang et al., 2020 |
|
SudarshanaChurna (Ayur.) |
Fever, Infection |
Naive Bayes + ADME |
COX-2, TLR4, IL-6 |
Multi-herb synergism predicted |
Soni et al., 2023 |
|
Licorice-Ginger-Clove |
Type 2 Diabetes |
CNN + Mol. Docking |
DPP-4, GLP-1R, PPAR |
Glabridin, Gingerol synergy validated |
Patel et al., 2021 |
|
Xiaoyao San(TCM) |
Depression, Anxiety |
GCN + PPI Network |
5-HT2A, MAOA, BDNF |
Multi-pathway modulation predicted |
Liao et al., 2022 |
TCM = Traditional Chinese Medicine; Ayur. = Ayurveda; GNN = Graph Neural Network; RF = Random Forest; SVM = Support Vector Machine; CNN = Convolutional Neural Network; GCN = Graph Convolutional Network; PPI = Protein-Protein Interaction
AI-DRIVEN PHARMACOLOGICAL SYNERGY PREDICTION
Computational Synergy Scoring
One of the most clinically significant applications of AI in poly-herbal drug discovery is the prediction of pharmacological synergy between herb components. Traditional approaches to synergy assessment, such as the Chou-Talalay combination index method, are limited to pairwise interactions and require extensive experimental data.[68]AI-driven synergy prediction platforms such as DeepSynergy[69] and SynergyX[70] employ deep neural networks trained on large combinatorial drug interaction datasets (e.g., NCI ALMANAC, ONEIL) to predict synergy scores for novel compound combinations. Applied to herbal phytochemical combinations, these tools can screen thousands of compound pairs from a poly-herbal formulation to identify the most synergistic bioactive pairs — dramatically reducing the experimental burden of synergy characterization.Network-based synergy prediction leverages the topology of PPI networks to identify compound combinations that co-modulate critical network hubs or act on proximal nodes in disease-relevant pathways.[71] The concept of 'network proximity' — measuring the shortest path distance between the target sets of two compounds in the interactome — provides a quantitative index of potential synergy that has been validated across multiple disease contexts.
Molecular Dynamics and Allosteric Synergy
Beyond network-level synergy, AI has been applied to predict allosteric synergy — where one compound modulates the binding affinity of another at the same protein target through conformational changes. Molecular Dynamics (MD) simulations, enhanced by ML force fields, can characterize allosteric communication pathways within target proteins.[72]Machine learning potentials (MLPs) such as ANI and SchNet[73] dramatically accelerate MD simulations while maintaining quantum mechanical accuracy, enabling microsecond-scale MD studies of herb compound-protein complexes that would be prohibitively expensive with classical force fields. These studies have revealed novel allosteric binding sites on established drug targets that may be exploited by minor herbal constituents acting as allosteric modulators.
MULTI-OMICS INTEGRATION IN AI-NP FRAMEWORKS
Transcriptomics and Gene Expression Data
The integration of transcriptomics data into AI-NP frameworks has substantially enriched the biological context of target network analysis.[74] Gene expression datasets from GEO (Gene Expression Omnibus) and ArrayExpress provide condition-specific profiles of disease-relevant gene regulation, which can be overlaid on compound-target networks to identify the subset of targets most likely to be pharmacologically accessible in the disease state.Connectivity Map (CMap) analysis, originally applied to drug repositioning, has been adapted for herbal pharmacology. By comparing the gene expression signatures induced by herbal treatments (derived from published transcriptomics studies) with CMap perturbagen signatures, researchers can infer the molecular mechanism of herb action and identify similarities with known pharmacological agents.[75]
Proteomics and Metabolomics
Proteomics data from PhosphoSitePlus and PhosphoELM enables integration of post-translational modification (PTM) networks into NP analysis, revealing how herbal compounds modulate kinase signalling cascades beyond simple binding interactions.[76] AI models trained on PTM data can predict how herbal compound-induced changes in kinase activity propagate through signalling networks, providing mechanistic depth beyond target binding prediction.Metabolomics data, when integrated with NP frameworks, creates a comprehensive metabolic NP approach in which metabolic network rewiring induced by herbal treatment is mapped alongside pharmacological target interactions.[77] This multi-layered framework is particularly relevant for poly-herbal formulations that modulate gut microbiome metabolism, producing secondary metabolites with distinct pharmacological properties from the parent plant compounds.
Pharmacogenomics and Personalized Herbal Medicine
The convergence of pharmacogenomics and AI-NP opens the possibility of genotype-guided poly-herbal prescription. Genetic variants in metabolizing enzymes (CYP2D6, CYP3A4, UGT1A1) and drug transporters profoundly influence herbal compound pharmacokinetics; AI models trained on pharmacogenomics-ADME interaction datasets can predict inter-individual variability in herbal drug response.[78]AI-assisted analysis of genome-wide association studies (GWAS) and single-nucleotide polymorphism (SNP) datasets, integrated with herbal compound target networks, can identify patient subpopulations most likely to benefit from specific poly-herbal formulations — a precision medicine approach to traditional medicine that is emerging as an exciting research frontier.[79]
KNOWLEDGE GRAPH-DRIVEN HERB-DISEASE NETWORKS
Construction of Herb Knowledge Graphs
Knowledge Graphs (KGs) provide a powerful framework for integrating heterogeneous biomedical information relevant to herbal pharmacology. A herb-oriented KG typically encompasses nodes representing: herbs, phytochemicals, proteins, diseases, pathways, clinical symptoms, and traditional medicine concepts; connected by typed edges encoding relationships such as 'contains', 'inhibits', 'associated_with', and 'treats'.[80]TCMKG[81] (TCM Knowledge Graph) and AyurKG[82] represent specialized KGs for TCM and Ayurveda respectively. These KGs, when queried using graph embedding methods (TransE, RotatE, ComplEx) or GNN-based link prediction models, can infer novel herb-disease associations and compound-target interactions beyond what is explicitly encoded in the graph.[83]
Natural Language Processing for Literature Mining
The biomedical literature contains an enormous volume of herb-compound-target-disease association data embedded in unstructured text. NLP-based information extraction pipelines, leveraging named entity recognition (NER), relation extraction (RE), and coreference resolution, enable automated construction and updating of herb KGs from PubMed literature.[84]
BioBERT[85] and PubMedBERT, transformer models pre-trained on biomedical text, achieve near-human performance on biomedical NER and RE benchmarks and have been applied to extraction of herb-compound-target triples from literature. HERB-KG projects integrating these NLP pipelines with curated databases are creating dynamic, auto-updating knowledge resources that remain current with the rapidly expanding herbal pharmacology literature.[86]
REGULATORY AND TRANSLATIONAL ASPECTS
Regulatory Landscape for AI-Designed Herbal Products
The regulatory pathway for AI-NP-guided poly-herbal formulations presents unique challenges at the intersection of traditional medicine regulation, computational drug discovery, and novel therapeutic entities.[87] Regulatory agencies including the US FDA, European EMA, and India's CDSCO have not yet established specific guidelines for the computational characterization of herbal multi-component preparations, creating a translational gap between AI-NP discoveries and regulatory approval.The WHO Traditional Medicine Strategy 2019–2025[88] and the FDA's Botanical Drug Development guidance provide partial frameworks for herbal product development. AI-NP studies can contribute to regulatory submissions by providing molecular mechanistic evidence supporting claimed therapeutic indications and identifying potential safety liabilities through in silico toxicology screening.
Validation Pipeline from In Silico to Clinical
Robust validation of AI-NP predictions requires a hierarchical experimental pipeline: (a) in vitro target binding assays (SPR, ITC, fluorescence polarization) for predicted DTIs; (b) cell-based functional assays confirming target engagement and downstream pathway modulation; (c) in vivo pharmacological studies in relevant animal models; and (d) clinical pharmacokinetic studies establishing human exposure profiles.[89]Biomarker identification through AI-NP can guide the design of clinical trials for poly-herbal formulations by identifying surrogate endpoints (protein biomarkers, metabolomic signatures) that reflect target engagement and mechanistic pharmacological activity.[90] This biomarker-guided approach is essential for demonstrating the scientific rigor of traditional medicine modernization to regulatory bodies and the broader scientific community.
CHALLENGES AND LIMITATIONS
Data Quality and Standardization
The quality and completeness of existing herbal databases remains a significant limiting factor for AI model performance. Many databases contain inconsistencies in compound nomenclature, incomplete structure-activity annotations, and taxonomic ambiguities in plant species identification.[91] Standardization initiatives such as the NAPRALERT project and WHO Monographs on Selected Medicinal Plants are addressing these issues, but substantial curation work remains.The chemical complexity of crude herbal extracts — containing hundreds to thousands of phytochemicals — means that database-curated compound lists represent an incomplete snapshot of the actual therapeutic moiety. AI models trained on this incomplete data may miss key bioactive components or attribute activity to database-present compounds at the expense of uncharacterized minor constituents.[92]
Interpretability and Explainability of AI Models
Deep learning models, while achieving high predictive performance, are often criticized as 'black boxes' with limited mechanistic interpretability. In the context of drug discovery, where understanding the molecular basis of predictions is essential for hypothesis generation and experimental design, this lak of interpretability is a critical limitation.[93]Explainable AI (XAI) approaches including SHAP (SHapley Additive exPlanations) values, LIME (Local Interpretable Model-agnostic Explanations), integrated gradients, and attention visualization are increasingly applied to drug discovery AI models to provide post-hoc explanations of predictions.[94] For herbal NP applications, XAI tools can highlight the molecular features (functional groups, pharmacophoric elements) most responsible for target binding predictions, guiding medicinal chemistry optimization of herbal leads.
Experimental Validation Gap
A persistent challenge in AI-NP herbal drug discovery is the gap between computational predictions and experimental validation. Publication bias toward successful predictions, combined with the high cost and time requirements of experimental validation, means that many AI-NP predictions remain computationally confined without biological confirmation.[95]
Systematic benchmarking studies comparing AI-NP prediction accuracy against experimental DTI datasets are essential for establishing confidence in these methodologies.
Summary of Challenges and Solutions
Table 3 provides a comprehensive overview of major challenges in AI-integrated network pharmacology for poly-herbal drug discovery, along with their implications and proposed AI-driven solutions
Table 3. Key Challenges in AI-NP-Based Poly-Herbal Drug Discovery and Proposed Solutions
|
Challenge |
Implication |
Proposed AI-Driven Solution |
|
Chemical complexity of herbal extracts |
Incomplete compound profiling |
LC-MS/NMR metabolomics + AI-assisted dereplication |
|
Lack of standardized databases for polyherbal formulations |
Data incompleteness, bias in ML training |
Development of curated ontologies (e.g., HERB, TCMBank) |
|
Polypharmacology and multi-target complexity |
Difficulty in isolating individual contributions |
Hypergraph and multi-layer network models |
|
Poor ADME/T prediction for plant phytochemicals |
High attrition in preclinical stages |
Transfer learning models trained on plant chemical space |
|
Synergy vs. antagonism identification |
Unpredictable drug interactions |
AI-based drug combination scoring (DeepSynergy, etc.) |
|
Regulatory validation gaps |
Limited clinical translation |
Adaptive trial designs guided by AI-predicted biomarkers |
|
Reproducibility of in silico findings |
Low external validity |
Cross-validation with public bioassay datasets (ChEMBL, PubChem) |
|
Black-box nature of deep learning models |
Low interpretability/trust |
Explainable AI (XAI) with SHAP/LIME integration |
XAI = Explainable Artificial Intelligence; SHAP = SHapley Additive exPlanations; ADME/T = Absorption, Distribution, Metabolism, Excretion, Toxicity; ML = Machine Learnin
DIRECTIONS
Digital Twins for Herbal Medicine
The concept of a 'Digital Twin' — a computational replica of a biological system that can simulate its responses to interventions — is emerging as a transformative paradigm in precision medicine. Applied to herbal pharmacology, a digital twin of a poly-herbal formulation would encode all known compound-target-pathway interactions and simulate the formulation's pharmacodynamic effects on virtual patient models parameterized with individual omics and clinical data.[96]AI-driven digital twins for herbal medicine could enable: (a) in silico optimization of formulation composition for specific patient phenotypes; (b) prediction of inter-individual variability in therapeutic response; (c) identification of herb-drug interactions for patients on concomitant conventional medications; and (d) simulation of dose-response relationships to support dosing optimization.[97]
Multimodal Foundation Models
Foundation models — large-scale AI models pre-trained on broad corpora and fine-tuned for specific tasks — represent the next evolution in AI-driven drug discovery. Models such as ESM-2 (protein language model)[98] and MolFM (molecular foundation model)[99] are pre-trained on comprehensive molecular and biological data and can be fine-tuned on herbal compound datasets with minimal labeled data, overcoming the data scarcity challenge that has historically limited AI applications in this domain.
Multimodal foundation models that jointly encode molecular structures, protein sequences, clinical phenotypes, and traditional medicine textual knowledge within a unified representation space hold promise for holistic AI-NP analysis that bridges biochemical, physiological, and ethnopharmacological knowledge dimensions.[100]
AI-Guided Microbiome-Herb Interaction Studies
Emerging evidence demonstrates that the therapeutic efficacy of many herbal formulations is substantially mediated by gut microbiome transformation of ingested phytochemicals into bioactive metabolites.[101] Berberine, for example, undergoes extensive microbial biotransformation producing dihydroberberine, which has superior glucose-lowering activity compared to the parent compound. AI models integrating microbiome metabolic pathway databases with NP frameworks can predict microbiome-dependent phytochemical transformations and their pharmacological consequences.This microbiome-NP-AI nexus represents an exciting emerging frontier with potential to explain inter-individual variation in herbal drug response attributable to microbiome composition differences, and to design synbiotic (herb + probiotic) combinations that optimize therapeutic efficacy.[102]
AI in Clinical Trial Design for Herbal Medicines
AI-NP has significant potential to rationalize clinical trial design for poly-herbal formulations. Network-derived biomarkers can define measurable outcome indicators aligned with the predicted mechanism of action, supporting adaptive trial designs with interim analyses based on biomarker response.[103] Patient stratification algorithms derived from NP-pharmacogenomics analyses can identify responder subpopulations, enabling enrichment strategies that improve trial efficiency and success probability.AI-assisted literature synthesis and meta-analysis of existing herbal clinical trial data can identify patterns of efficacy and safety that guide hypothesis generation for prospective trials. NLP tools applied to published RCT reports and traditional medicine clinical literature can extract structured efficacy and safety data, supporting systematic reviews and network meta-analyses that integrate evidence across diverse traditional systems.[104]
Quantum Computing Perspectives
Quantum computing, while still in early development stages, holds long-term promise for accelerating computational drug discovery applications.[105] Quantum algorithms for molecular simulation (variational quantum eigensolver, quantum phase estimation) could eventually enable exact calculation of binding free energies for herb compound-target complexes — currently the principal source of inaccuracy in computational docking approaches. Hybrid quantum-classical algorithms combined with AI are actively being explored for drug-target interaction prediction and molecular property optimization.[106]
13. ETHICAL AND INTELLECTUAL PROPERTY CONSIDERATIONS
The application of AI to traditional herbal medicine raises important ethical considerations regarding intellectual property, cultural heritage, and equitable benefit sharing. Traditional knowledge encoded in Ayurveda, TCM, Unani, and Siddha systems represents the intellectual heritage of indigenous communities and nations.[107] The Convention on Biological Diversity (CBD) and the Nagoya Protocol establish frameworks for access and benefit sharing (ABS) that must be respected in commercial applications of AI-derived insights from traditional medicine databases.The Traditional Knowledge Digital Library (TKDL) established by the Government of India serves as a defensive publication tool to prevent misappropriation of traditional knowledge through patent systems. AI-NP researchers should engage with these frameworks proactively, ensuring that commercial development of AI-guided herbal formulations benefits the traditional knowledge communities whose empirical insights underpin these discoveries.[108]Additionally, the training of AI models on traditional medicine textual databases raises concerns about the accuracy and cultural sensitivity of extracted knowledge. Traditional medicine texts were authored within specific cultural-philosophical frameworks that may not map cleanly onto modern biomedical concepts; AI models must be designed with this contextual awareness to avoid misrepresentation or decontextualization of traditional knowledge.[109]
CONCLUSIONS
The integration of Artificial Intelligence with Network Pharmacology represents a scientifically rigorous, computationally powerful, and practically translatable approach to poly-herbal drug discovery. This review has documented the substantial advances achieved in recent years: from classical ML-based target prediction to GNN-driven DTI models, from manual network construction to automated knowledge graph inference, and from individual herb analysis to whole-formulation synergy prediction.Key strengths of this integrated approach include: scalability to handle the chemical and biological complexity of multi-herb formulations; the capacity to generate mechanistic hypotheses amenable to experimental testing; the ability to identify novel pharmacological utility for traditional formulations; and the potential to guide clinical translation through biomarker identification and patient stratification.Despite significant progress, critical challenges remain: data quality and completeness, model interpretability, experimental validation gaps, and regulatory uncertainty. Addressing these challenges will require coordinated efforts from computational scientists, pharmacologists, clinical researchers, regulatory agencies, and traditional medicine practitioners.Looking forward, the convergence of AI-NP with multi-omics, digital twins, microbiome science, federated learning, and quantum computing promises to further deepen and accelerate herbal drug discovery. As these technologies mature, the ancient wisdom embedded in global traditional medicine systems will increasingly serve as a validated, evidence-based source of novel therapeutic leads and poly-pharmacological strategies for complex diseases of the 21st century.
REFERENCES





Sadiya Prabin, K. Hamsika Sri, Edalada Pavan Kumar, Gurleen Kaur, Dr. Md Sayeed Anwar, AI Integration with Network Pharmacology for Poly Herbal Drug Discovery: Current Advances and Future Scope, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5234-5256, https://doi.org/10.5281/zenodo.20772710
 10.5281/zenodo.20772710
10.5281/zenodo.20772710