
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
St. Soldier Institute of Pharmacy, Lidhran Campus Behind NIT (R.E.C) Jalandhar- Amritsar byepass, NH-1, Jalandhar- 144011, Punjab, India.
Fibrodysplasia Ossificans Progressiva (FOP), commonly known as Stone Man Disease, is an exceptionally rare and disabling genetic disorder characterized by progressive heterotopic ossification, in which bone forms within muscles, tendons, ligaments, and other connective tissues. The disorder is primarily caused by gain-of-function mutations in the ACVR1 gene, leading to abnormal activation of bone morphogenetic protein (BMP) signaling pathways and irreversible ectopic bone formation. The disease usually manifests during early childhood with congenital malformations of the great toes, followed by recurrent inflammatory flare-ups that progressively restrict joint mobility and result in severe physical disability. Diagnosis is mainly based on characteristic clinical features, radiological findings, and confirmation through genetic testing. At present, no definitive cure is available, and management is focused on preventing trauma, controlling inflammation, relieving pain, and preserving quality of life. Emerging therapeutic strategies, including ACVR1-targeted therapies, activin A inhibitors, immunotherapy, nanoparticle-based drug delivery systems, and gene-targeted interventions, are currently under investigation and offer promising future treatment possibilities. In addition, selected herbal compounds with anti-inflammatory and antioxidant properties, such as curcumin, ginger, Boswellia, Ashwagandha, and Ginkgo biloba, have been explored as supportive therapies, although robust clinical evidence remains limited. This review summarizes the epidemiology, pathophysiology, clinical manifestations, diagnosis, current therapeutic approaches, novel treatment strategies, and recent advances in FOP research. A comprehensive understanding of the molecular mechanisms underlying the disease may facilitate earlier diagnosis and support the development of targeted therapies capable of improving long-term outcomes and quality of life for affected individuals.
An uncommon hereditary condition called fibrodysplasia ossificans progressive (FOP), formerly known as myositis ossificans progressive is characterized by extensive regions of progressive heterotopic endochondral ossification (HEO). Incidence is approximately one per 2 million people worldwide. The growth of an additional hard skeleton around the normal skeleton causes FOP, a terrible illness known as "Stone man disease," to have significant cumulative impairment. The disease is rarely identified in these kids at birth, despite the fact that they have some telltale symptoms such bilaterally undersized great toes with hallux valgus and aberrant calcaneal ossification. Hard bone masses grow in muscles, ligaments, aponeuroses, and joint capsules after the symptoms, which usually start in the first ten years of life with recurrent episodes of excruciating inflammatory soft tissue swellings. By the third decade of life, a patient's quality of life and everyday activities are severely limited due to a progressive loss of motion at numerous joints. The present choices for treatment are limited to palliative and symptomatic treatments as there is no known definitive cure for the disease.
Case report of a 10-year-old child who had multiple bony masses in different parts of the body and severe upper limb contractures due to joint contractures. The child never complained of painful soft tissue lesions or disease flare-ups, even after being questioned about the incident repeatedly [1]. Patin provided the first description of it. Its physiopathology is yet not fully known. odema affects the soft tissues, with the back of the neck being the most commonly afflicted area. This edema, which results from inflammatory processes, calcifies over time and causes the affected area to lose mobility. Trauma that occurs again may start the process.
Numerous therapies, including oral etidronate, isotretinoin, and steroids, have been tried, but with mixed success. According to in vitro chemical research, bisphosphonates reduce the
production of heterotopic bone during the active stage of the disease by adsorbing hydroxyapatite crystals. High ascorbic acid dosages, according to Palhares' observations,
slowed the disease's progression [2].
The two main features of FOP are increasing heterotopic ossification in certain anatomic patterns that results in early impairment and congenital skeletal abnormalities of the great toes. Even though genetic transmission is autosomal dominant, spontaneous novel mutations account for the majority of sporadic instances. Skeletal components merge with regular skeletal bone during childhood as new bone forms within soft connective tissue, such as skeletal muscle, tendon, and ligaments. Inflamed and swollen soft tissue occurs prior to ossification. Heterotopic bone development is sporadic, with ossification typically following a set pattern that begins in the neck and upper back. This condition is progressive leading to an early handicap due to contractures and joint fusions.
Any mild trauma, including intramuscular injections, or soft tissue injury might hasten the disease's natural progression. Additionally, blunt muscle damage from bumps, bruises, and falls, as well as mandibular blocks used for dental procedures, muscular exhaustion, and immobilization may encourage the production of heterotopic bone at the site of injury. Attempts to remove heterotopic bone surgically or by biopsy run the danger of inducing abrupt and excruciating development of new bone3. For FOP, there is currently no proven effective therapy. Although there is little concrete data to support it, radiotherapy treatment may have some therapeutic benefit for patients with FOP. Additionally, there is a dearth of literature on the subject of radiotherapy treatment for FOP patients [3].
1.1. HISTORY
Previous natural history studies on flare-ups (FLOP) were carried out before the period of corticosteroid symptomatic treatment, more than 20 years ago, and analyzed a small number
of participants (about 40). Furthermore, because the FOP community was small and regional, these early investigations were neither worldwide nor comprehensive. The organized FOP community has expanded significantly over the last 20 years, partly because of the internet's and social media's explosive expansion as well as the efforts of concerned patients, physicians, and families [4]. Medical records from Dr. Guy Patin in 1692 describe people who suffer from FOP. Myositis ossificans progressive, the original name for FOP, was believed to be the result of muscle inflammation (myositis) that led to the production of bones. Victor A. McKusick renamed the condition in 1970 after it was found that the disease process also damaged soft tissue, such as ligaments, in addition to muscles [5].
The FOP case involving Harry Eastlack (1933–1973) is the most well-known. His illness started when he was ten years old, and by the time he passed away from pneumonia in November 1973—six days before turning forty—his body had fully ossified, limiting his range of motion to his lips[6].
But Eastlack gave science a gracious donation of his corpse, and his skeleton is currently housed at the Mütter Museum in Philadelphia, where it is an invaluable tool for researching FOP. In February 2019, the skeleton of Carol Orzel (April 20, 1959 – February 2018), a second person with FOP, was placed next to Eastlack's in the museum[7].

Fig:1 :- Stoneman Disease
2. EPIDEMIOLOGY
As of 2017, there were only about 800 cases of FOP confirmed worldwide, making it an extremely rare condition. Because of its rarity, FOP is regarded as one of the most unusual illnesses in human history. FOP affects individuals of all ethnicities and has an estimated incidence of 0.5 cases per million, despite its low prevalence. It's crucial to remember that these figures are still valid and applicable today[8].
About one in every two million persons worldwide are afflicted with FOP, an extremely rare genetic disorder. It impacts every geophysical predisposition and shows no respect for gender, race, or ethnicity. shows how diverse autosomal dominant trait transmission occurs expression when it occurs frequently and intermittently. The condition appears early in life and progresses postnatally. Life must go inexorably [9].
The Japanese Ministry of Health, Labour, and Welfare estimates that 500,000 individuals globally are impacted. FOP does not support a particular race, ethnicity, gender, or geographic area. FOP is extremely rare, occurring about once every two million people worldwide. There has been no mention of any racial, ethnic, or geographic predispositions. One of the rarest diseases ever discovered, FOP had approximately 800 confirmed cases worldwide as of 2017. The estimated 0.5 instances of FOP per million population affect all ethnic groups [10].
3. PATHOPHYSIOLOGY
ALK2 transduction is triggered by ligand interaction and is caused by a gain of function mutation in the ACVR1/ALK2 gene, which codes for ALK2 kinases. In [11][12] One of the type II receptors, such as BMP receptor type II (BMPR-II), activin receptor type IIA (Acta-IIA), and activin receptor type IIB (Acta-IIB), and the TGF-β family ligands, such as BMP-6, BMP-7, BMP9, and activin B, bind to the extracellular domain of ALK2, a type I receptor[13].
As constitutively active ser/thr kinases, these type II receptors cause phosphorylation of ALK2 at the GS domain, which is made up of serine and glycine residues in the ternary complex that is created when the ligand binds to ALK2 at the cell membrane. As a result of this phosphorylation of ALK2, downstream substrates like SMAD1, SMAD5, and Smad8/9 have their serine and threonine residues phosphorylated, activating the kinase activity [14][15].When these phosphorylated Smad proteins attach to particular DNA sequences, they control the target genes' transcription in the nucleus, which causes the heterotopic ossification that fibrodysplasia ossificans progressiva patients experience. As a result, all of these mutations cause intracellular signaling, in which ALK2 collaborates with type II receptors to function as receptors[16][17].
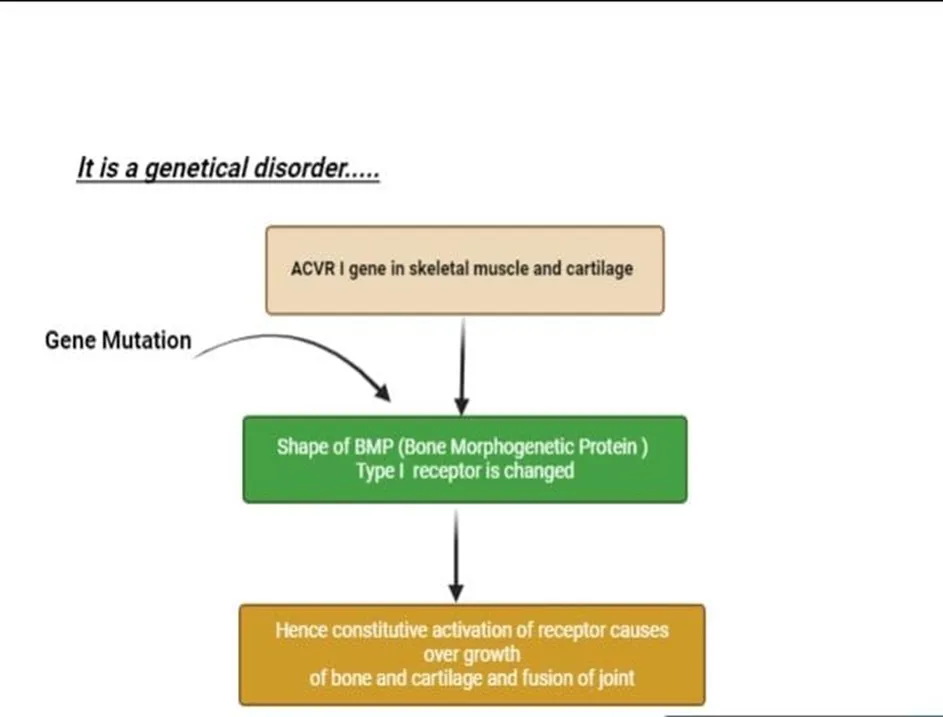
Fig:2 :- PATHOPHYSIOLOGY OF STONEMAN DISEASE
4. CASE PRESENTATION
A 10-year-old child from Pakistan came to our clinic in Pakistan six months ago complaining of pain and sore spots on his left arm, left hip, and back. The pain started slowly and got worse over time, making it harder to walk and limiting shoulder and hip range of motion. Upon examination, the patient presented with several swellings on his left hip, left knee, right shoulder, and back. On the left arm, close to the anterior fold of the armpit and extending to the entire biceps, there was another firm lump visible. There was no warmth or inflammation visible, yet the mass hurt (Fig. 3). Upon palpation, all visible masses were found to be painful, and all abdominal and paraspinal muscles were tight. There was a limit of 35 degrees on the left side and 10 degrees on the right side for abduction of both shoulders (Fig. 3).
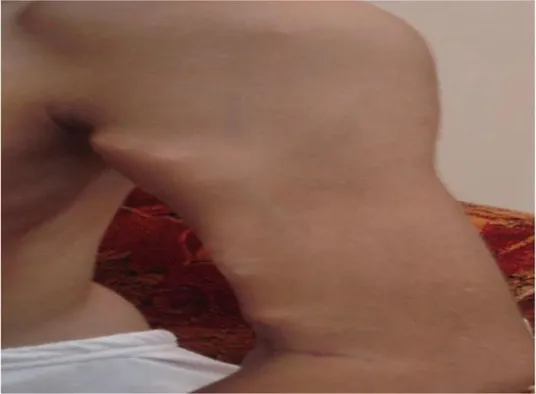
(Fig. 3):- Disease present in left shoulder
mass on the left arm and a restricted 35-degree angle in the abduction of the shoulders The patient did not have any other toe abnormalities other than bilateral hallux valgus
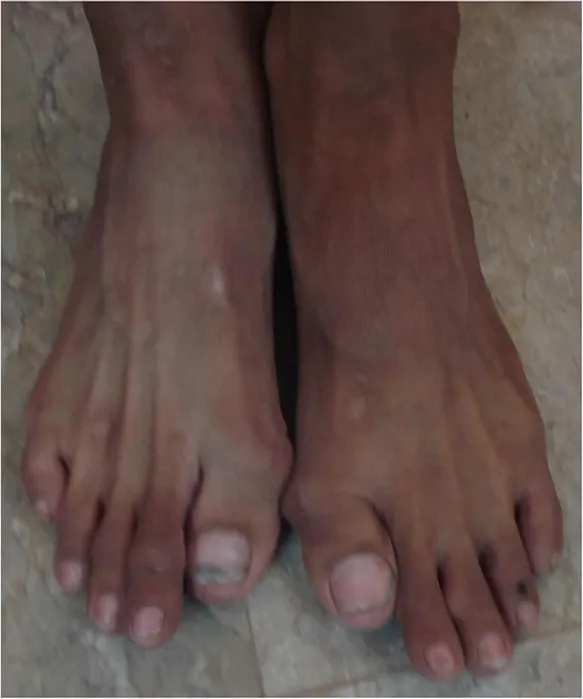
Fig:-4 :- Disease present in feet
He had severely limited range of motion in his left hip and was unable to walk or squat in a normal posture (Fig. 5).
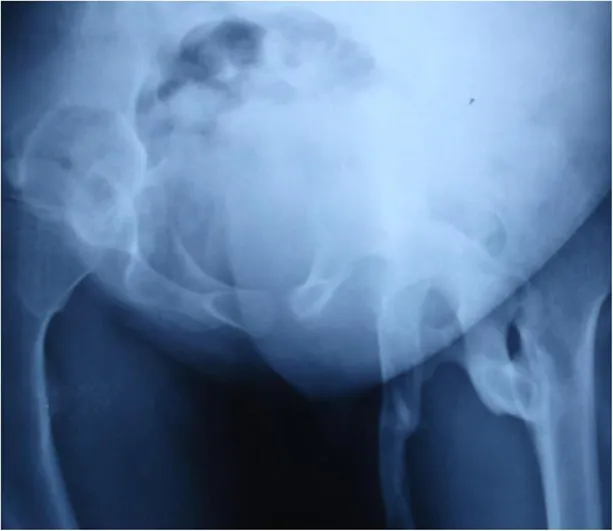
Fig:5 :- Abnormalties occur in hip bone
There was no discernible lymphadenopathy. When the boy's parents examined him physically, they found no comparable anomalies.
Laboratory study results were within normal limits. The parents' financial difficulties prevented the performance of genetic analysis testing. Heterotopic ossification including the right knee, hips, shoulders, neck, and spine was visible on conventional radiography (Figs.6 and 7).
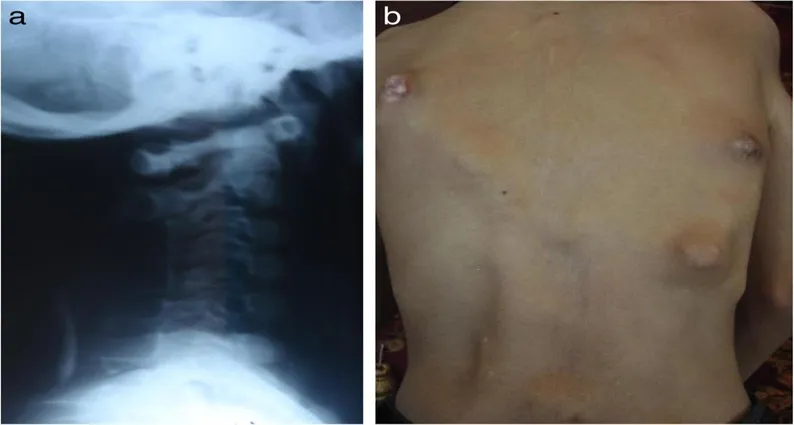
Fig :6 Abnormalaties Occur in Spine and Neck
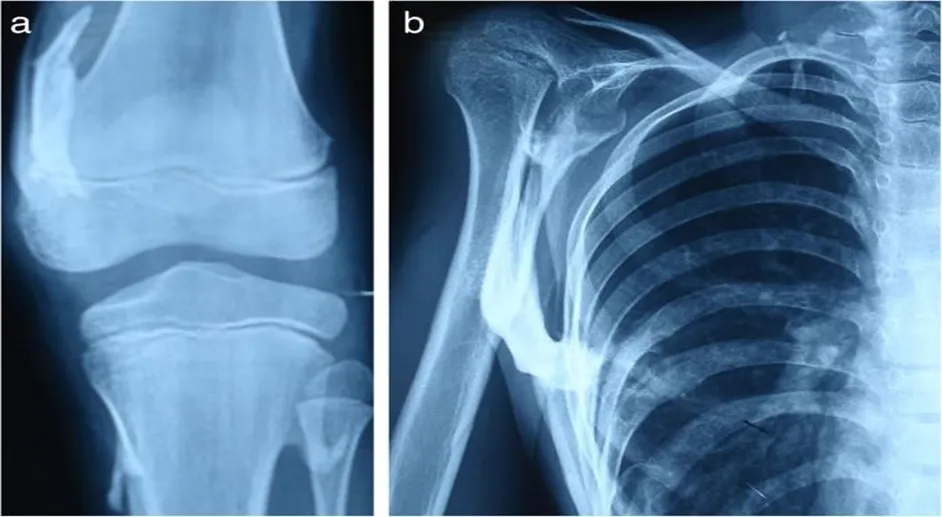
Fig :7 :- Abnormalaties occur in Knee and Shoulder
At the start of the illness, there was no history of local trauma. The patient didn't have any siblings or a medical history in her family.
Concerning the patient's prior medical history, his parents stated that a surgical procedure resulted in the excision of a left hip ossification, which was followed by a brief improvement in the hip's range of motion. Periodic evaluations were conducted every two months, and the results of the evaluation revealed a flare-up (aggravation) of the ossification and substantial restriction in the left hip's range of motion. (Fig. 8).
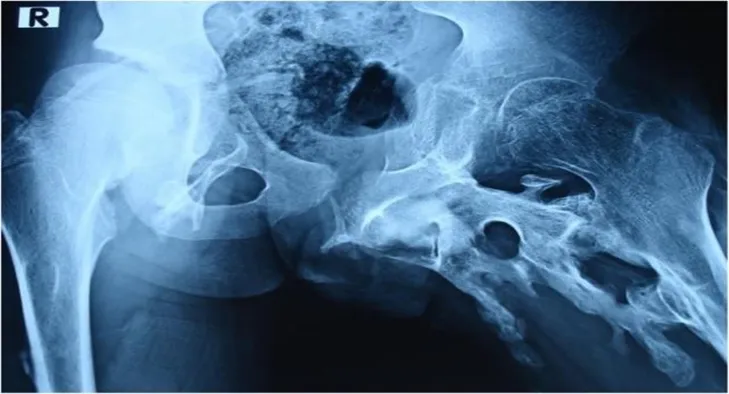
Fig:8 :- Disease occur in left hip
X-ray follow-up following surgery or excision: Right hip ossification is present and equivalent to the preoperative x-ray; however, the left hip ossification has flared up excessively.
The patient was started on symptomatic treatment and his family was informed about the condition.
Counseling was provided, and it was suggested that minor trauma be avoided. It was noted at a recent follow-up that he was living a better quality of life than when he was last there. The patient will have radiological and clinical monitoring[18].
5. OTHER NAMES [19]
Fig :9 :- Other name of stoneman Disease
6. Symptoms & Signs :
• Swelling similar to a tumor in the shoulder and other joints; • Fusion of the toe bone
• Thumb and little toe
• Pain and fever
• Swellings in tendons
• A progressive neck fusion
• Scoliosis, or abnormally curved spine on the side
• Ossification of fibrous and muscle tissue
• Limited range of motion in the neck, spine, wrist, shoulder, and knee
• Eating disorders
• Having trouble speaking
• Congestive cardiac failure of the right side
• A viral respiratory illness
• Minimal intellectual impairment
• Having trouble sitting and walking
7.Therapeutic approaches
As of right now, HO in FOP has no proven medical treatment. The primary therapeutic strategy is to prevent trauma or injury to the muscles. Growth and recurrence following ossification removal surgery are common outcomes. For spinal abnormalities, bracing is
ineffective; nevertheless, activity restriction can lessen damage. Occupational therapy and career education consultations are beneficial [20].
Temporary relief from flare-up symptoms can be achieved with high-dose corticosteroids; recurrent flare-ups can be treated with non-steroidal anti-inflammatory drugs (NSAIDs), cyclooxygenase 2 (COX2) inhibitors, mast cell inhibitors, and muscle relaxants. Potential therapy involves blocking the ACVR1/ALK2 gene-related pathway, which stops aberrant
bone growth. Medication like rapamycin and palovarotene are undergoing clinical trials, and surgery is not usually the first choice for FOP patients [21]. A number of drugs, such as rapamycin and palovarotene, are presently under clinical studies to treat Stoneman syndrome. For some FOP patients who also have coexisting diseases, these drugs do have certain constraints and limitations. For example, the known teratogen palovarotene can cause limb abnormalities in the growing fetus and may harm the child's growth plates, hearing, and vision. Although most people tolerate the medicine well, mucocutaneous side effects are the main cause for concern. However, there are hazards associated with rapamycin use in kidney transplant patients, including dyslipidemia, proteinuria, and oral ulcers [22].Patients who had rapamycin treatment for vascular anomalies reported toxicity in their bone marrow, blood, and digestive tract. Rapamycin has a black-box warning that advises against using it in individuals with liver disease because of a higher risk of infection andimmunosuppression [23].
These difficulties add to the difficulty of creating treatments for FOP. There are various challenges in the way of developing new therapeutic targets for FOP. These difficulties are exacerbated by the disease's rarity and heterogeneity, our incomplete knowledge of its underlying mechanisms, the scarcity of appropriate animal models, worries about safety, and the possibility of flare-ups [24].Exaggerated inflammatory response is a hallmark of FOP, and any trauma or tissue injury can set off flare-ups that lead to the production of aberrant bone. There are few reliable animal
models that accurately mimic the features of FOP, and the disease's incomplete understanding makes it difficult to identify specific targets for therapeutic intervention [25].
Using therapeutic procedures, including surgery or medication therapies, is challenging due to the possibility of making the disease worse [26].
Finding new therapeutic targets for FOP could change the course of the condition, enhance symptom management, personalize treatment plans, lessen adverse effects, and advance our understanding of bone biology. These potential advantages give optimism for individuals .
8. Other aproach
8.1Immunotherapy
, which targets specific cells or molecules with the immune system, is a growing area of research in FOP. Researchers are investigating a number of
immunotherapeutic approaches, including immune checkpoint inhibitors and monoclonal antibodies, that can precisely target the cells causing abnormal bone development. By controlling the immune system, these therapies aim to avoid or minimize HO in FOP [27-28]. Proteins known as monoclonal antibodies have the ability to bind to specific antigens on the surface of cells, triggering the immune system to begin eliminating those cells. Immune checkpoint inhibitors are drugs that can mute the immune system's signaling pathways, preventing it from attacking healthy cells. By using these immunotherapeutic drugs,
researchers hope to enhance the immune system's capacity to identify and eliminate the aberrant cells that cause FOP [29-30].
8.2 Nanoparticle Delivery
Therapeutic medications may be delivered by nanoparticles to the affected tissues in FOP. Researchers are looking at the use of nanoparticles to deliver tiny medications, gene-editing instruments, and other therapeutic therapies specifically to areas of aberrant bone
development. This tailored delivery approach aims to minimize side effects and increase the effectiveness of potential FOP treatments [31].
Nanoparticles are minuscule particles that can be produced and have specific properties like size, shape, charge, and surface chemistry. Moreover, drugs, genes, or proteins can be added
to nanoparticles to make them therapeutic. It is possible to design nanoparticles such they can cross biological barriers, such as the skin's barrier or the blood-brain barrier, and get to the
intended areas. Additionally, targeting ligands, such as peptides or antibodies, that can recognize and bind to particular receptors on target cells can be designed into nanoparticles [32–33].
9. Research being done now on new therapeutic targets
Even though there is a lot of promise in finding new therapeutic targets for FOP, most of the current research is preclinical. Scientists are using in vitro studies and animal models to do extensive study on the safety and effectiveness of these emerging approaches. Numerous preclinical studies have looked into potential FOP treatments by examining the molecular mechanisms of HO and the effects of ACVR1 mutations. Promising targets include BMPs,
senescence, hypoxia, inflammation, and activin A. A ligand called activin A attaches itself to ACVR1 and causes HO in FOP. In animal models of FOP, inhibiting Activin A signaling with monoclonal antibodies or small molecules has been reported to decrease HO [34].
Ligands known as BMPs bind to ACVR1 and control the growth of new bone. It has been shown that BMP signaling inhibition by antibodies, small molecules, or gene therapy can prevent or correct HO in animal models of frontotemporal palsy [35].
Low oxygen levels, or hypoxia, are what cause HO to occur in FOP. In animal models of FOP, addressing hypoxia-inducible factors (HIFs) or downstream pathways with medications or gene therapy has been found to reduce HO [36].
Inflammation induces HO in FOP as a reaction to tissue damage or infection. In animal models of FOP, it has been demonstrated that modifying inflammatory cytokines,
chemokines, or receptors using medications or gene therapy attenuate
10. Herbal Treatments
1. Turmeric/Curcumin
Curcumin, a polyphenol in turmeric, has anti-inflammatory and antioxidant properties that may help alleviate FOP symptoms [37].
2. Ginger
Ginger's anti-inflammatory compounds, gingerol and shogaol, may help reduce pain and inflammation in FOP patients [38].
3. Boswellia
Boswellia's anti-inflammatory properties may help reduce pain and inflammation in FOP patients [39].
4. Ashwagandha
Ashwagandha's adaptogenic properties may help reduce stress and anxiety in FOP patients [40].
5. Ginkgo Biloba
Ginkgo biloba's anti-inflammatory and antioxidant properties may help improve circulation and reduce inflammation in FOP patients [41].
Important Notes
- These herbal treatments are not a replacement for conventional medical treatment. Consult a healthcare professional before using them.
- FOP is a rare disease, and more research is needed to confirm the effectiveness of these herbal treatments.
11.Drugs used in stoneman disease
Approved Treatments:
1. Prednisone (Glucocorticoid) - inflammation and flare-ups
2. Ibuprofen (NSAID) - pain management
3. Naproxen (NSAID) - pain management
12.Diagnosis
-Based on clinical findings, the deformity of the toes is the hallmark of FOP, whereas other developmental anomalies are primarily restricted to the cervical spine and thumb. One of the main abnormalities of FOP is stiffness of the neck. The spine mostly forms huge vertical bodies through the union of C2 and C7[42][43][44] . The majority of individuals have early-stage ankylosis of the spine. Many individuals experience flare-ups, fusions, and other hallmarks of FOP, which can lead to the development of arthritic symptoms. Short malformed thumbs, clinodactyly, short broad femoral necks, and proximal medial tibial osteochondromas are a few more symptoms of FOP patients [44][45].-Radiographic characteristics of FOP: X-rays and bone scans are unable to identify the disease in its early stages MRI and CT scans can reveal lesions to a certain degree. However, from a diagnostic or general perspective, radiographic data doesn't provide much proof of the illness. Therefore, it is better to follow the clinical results [46][47].-FOP Laboratory Parameters: The ankylosis of the skeletal tissues causes a rise in alkaline phosphate in the blood. When there is a flare-up, the ESR rate rises [42] .
For tracking the acute inflammatory phase, C-reactive protein is far better than ESR rate; however, there is no evidence to support this claim [43]. Because of the additional bone and flare-up, urinary parameters such fibroblast growth factors are also elevated [42]. During the fibroproliferative, chondrogenic, and osteogenic stages of flare-ups, there may be a rise in cartilage-derived retinoic acid protein (CD-RAP) [48].
-Genetic testing: A diagnosis of FOP before the onset of Heterotrophic Ossificans can be
confirmed by conclusive genetic testing of FOP using DNA sequence analysis [49]
-ACVR1 Mutational Analysis: Using genomic DNA derived from regular venipuncture, DNA sequence analysis and polymerase chain reaction (PCR) evaluation were carried out in accordance with conventional procedures. By employing primers that flank the recurrent c.617G>A mutation found in patients with typical FOP, genomic DNA was amplified in order to screen for mutations
InACVR1[49].
DISCUSSION
Imaging tests and clinical manifestations are typically used to diagnose FOP. However, genetic analysis is required as well [50].
The majority of documented FOP patients have the ACVR1 gene molecule (c.617G > A, p.R206H); those who have this classical molecule exhibit an early beginning of ossifications [51].
The illness typically progresses severely and causes immobilization early on [52].
In this patient population, the initial ectopic ossifications typically occur in childhood and are often characterized by spontaneous bone nodules [53].
Nearly every patient observed the big toe's congenital deformity [54] .
In addition to immobilizing all major joints, the progressive development of ectopic ossification causes respiratory dysfunction due to severe scoliosis; the majority of patients have trouble breathing in their third or fourth decade of life [55].
CONCLUSION
Fibrodysplasia Ossificans Progressiva (FOP) is an extremely rare and debilitating genetic disorder characterized by the abnormal formation of bone in soft tissues, a process known as heterotopic ossification.
As of 2017, fewer than 800 cases have been confirmed worldwide, with an estimated incidence of 0.5 cases per million people, affecting all ethnicities and genders. FOP typically presents in childhood, with the hallmark symptom being the congenital malformation of the big toe. Over time, the condition progresses, causing painful, spontaneous bone growth in muscles, tendons, and ligaments, leading to joint immobility and respiratory difficulties.
The underlying genetic mutation in the ACVR1/ALK2 gene causes a gain-of-function mutation, which triggers abnormal bone formation through the dysregulation of signaling pathways involving bone morphogenetic proteins (BMPs) and transforming growth factor-beta (TGF-β) family ligands.
Clinically, FOP presents with pain, swelling, and limited mobility in affected areas, often progressing to a severe form of ankylosis, particularly in the spine, resulting in a stiff, immobile neck and other skeletal deformities. Radiographic imaging, such as X-rays and MRI, may detect the lesions in later stages, but early diagnosis relies heavily on clinical observations and genetic testing. Inflammation during flare-ups is reflected by elevated markers like alkaline phosphatase, ESR, and C-reactive protein, while urinary markers may also rise during active phases.Genetic testing for mutations in the ACVR1 gene (c.617G > A, p.R206H) can confirm the diagnosis before heterotopic ossification becomes evident.
Unfortunately, despite advancements in understanding, there is currently no cure for FOP, and its progression often leads to severe disability, including respiratory complications in later life due to scoliosis and thoracic cage deformities.
REFERENCES
https://doi.org/10.1016/j.gene.2013.06.022
https://doi.org/10.1016/j.gene.2013.06.022
https://doi.org/10.1016/j.gene.2013.06.022

Ravroop Singh, Rajesh Kumar, Ajeet Pal Singh, Amar Pal Singh, Gaurav Hastir, Review Article on Stoneman Disease, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 2122-2136, https://doi.org/10.5281/zenodo.21294580
 10.5281/zenodo.21294580
10.5281/zenodo.21294580