
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Department of Pharmaceutical Quality Assurance, R. C. Patel Institute of Pharmacy, Karwand Naka, Shirpur, Dist. Dhule 425 405 (M. S.) India
2Department of Pharmaceutical Quality Assurance, Adarsh Shikshan Prasarak Mandal’s K. T. Patil College of Pharmacy, Osmanabad (Dharashiv)
The active pharmaceutical ingredients (APIs) are defined as "the biologically active component of a medicinal product which is subject to regulation for establishing quality, safety, and efficacy." The global API market size was USD 215 billion in 2023 due to India's contribution of 20-25 percent of the overall global generic API market size through product supply. The Central Drugs Standard Control Organisation (CDSCO) of India and the United States Food and Drug Administration (USFDA) are two of the most strategically important systems for the global API industry. The narrative review is based on a complete comparison study, which evaluates the systems of regulation for API registration and Drug Master File (DMF) submission in India, i.e., CDSCO, and the United States, i.e., USFDA. The review is based on both formats of submission, including institutional, structural, and legal systems, documentation, which includes drug substance starting material (DSSM) designation standards, GMP standards, approval, and change requirements, including regulatory issues and harmonization. Methods: The authors performed a narrative review which examined primary regulatory guidance documents and scientific literature. The researchers conducted a literature search through PubMed Scopus and Web of Science databases to find published works between January 2015 and March 2025 using these search terms 'API registration' 'Drug Master File' 'CDSCO regulatory' 'USFDA API' 'ICH guidelines pharmaceutical' 'Schedule M GMP' and 'eCTD submission. The researchers obtained regulatory guidance documents from official websites of CDSCO USFDA ICH WHO and PIC/S. The researchers verified all regulatory information by checking primary source documents which included documents that showed their version dates. Findings: The foundation of ICH quality guidelines, specifically ICH Q7 (API GMP), is shared by both regulatory systems [18].Pharmaceutical development (Q8) [19], Q9 (quality risk management) [17], Pharmaceutical Quality System (Q10) [20], Q13 (continuous manufacturing, 2023) [23] and Q11 (API development) [21].There are significant distinctions between the SUGAM portal hybrid system in India and the mandatory eCTD via the FDA Electronic Submissions Gateway since May 2017/May 2018 [29, 20]. (ii) DMF formalisation: India's less codified IDMF versus the FDA's five-type DMF system with more than 15,000 active DMFs [8,40];(iii) starting material designation: CDSCO's ICH Q11-recommended but less enforced method versus the FDA's strict DSSM Guidance 2022 [9,21]; (iv) inspection rigour: CDSCO/SDA joint inspections versus the required USFDA Pre-Approval Inspection [42]; (v) data integrity enforcement: According to an analysis of FDA enforcement records, data integrity violations were the most commonly cited observation category in Warning Letters to Indian facilities between 2014 and 2022 [7,43,44]. The USFDA strictly enforces ALCOA+. and (vi) user fees: nominal CDSCO regulatory fees versus significant GDUFA III fees (DMF ~USD 62,420; facility ~USD 388,137 in FY2024) [41]. In conclusion, India's PIC/S observer status (2021) reinforces ICH harmonisation [14] and the updated Schedule M (G.S.R. 228(E), March 28, 2023) [11] The regulatory differences between India and the USA will continue to decrease because both countries have established new regulatory frameworks. Indian API manufacturers must strategically invest in eCTD readiness, ICH Q11-aligned starting material justification, ALCOA+ data integrity systems, and proactive nitrosamine risk assessment to sustain market access in the USA.
The ICH Q7 standard defines active pharmaceutical ingredients APIs which are also known as drug substances as any substance or mixture of substances that manufacturers will use to create a drug product because it will become an essential component of that drug product during the manufacturing process. [18]. The APIs present in finished pharmaceutical formulations act as their active components which create the desired medicinal effects; the therapeutic effectiveness and safety of patients depend on the testing results which verify the APIs' quality and purity and identification and strength.
The global pharmaceutical market which reached a value of approximately USD 1.48 trillion in 2022 and is expected to grow to USD 2.1 trillion by 2030 will experience a compound annual growth rate of 4.8 percent. [50] and a large number of USFDA-approved pharmaceutical manufacturing sites which produce approximately 20 to 25 percent of the world's generic active pharmaceutical ingredients through their actual output. [1,2]. The commonly cited figure of '42% of APIs consumed in the United States from India' should be interpreted with caution because this estimate varies across different methods which assess either volume or value or approved API source count. [2].
The complete API development process from its initial development stage to its final pharmaceutical product stage requires companies to follow the comprehensive regulations which all international regulatory bodies have established. The worldwide regulatory system shows jurisdictional diversity because each major regulated market including the USA and European Union and Japan and Canada and Australia has developed its own legal system and submission process and inspection requirements while international regulatory bodies ICH and WHO work to reduce these variations. [3,4].
The USFDA through its CDER division and Office of Pharmaceutical Quality which started in 2015 operates one of the most demanding pharmaceutical regulation systems worldwide. The Federal Food Drug and Cosmetic Act FD&C Act together with its Title 21 Code of Federal Regulations 21 CFR regulations creates the legal framework for assessment. [5,6]. USFDA's mandatory eCTD submissions (since 2017–2018) [29,20], formal five-type DMF classification system [8], stringent DSSM designation standards [9], pre-approval inspection programs [42], and rigorous data integrity enforcement [7] establish global regulatory benchmarks.
India's CDSCO, operating under the Drugs and Cosmetics Act 1940 [10], modernization was heavily revised by the deployment of the SUGAM digital era portal. [13], the revised Schedule M 2023 (Gazette Notification G.S.R. 228(E), dated 28 March 2023) [11] incorporating ICH Q7 and ALCOA principles, PIC/S observer status (2021) [14], and CDSCO Data Integrity Guidelines 2022 [12]. Indian API manufacturers face difficulties in meeting USFDA requirements because they must ensure data integrity, achieve DSSM designation, conduct nitrosamine impurity testing, and submit compliant eCTD documents. [7,9,15,16].
1.1 Aims and Objectives
The narrative review examines API registration regulations from India and the United States through a structured comparative research framework. The study aims to achieve its specific objectives through this academic analysis.:
The review examines the process of small-molecule chemical API registration which occurs in India and the United States. The study treats biosimilars and ATMPs as biological APIs while mentioning veterinary APIs and controlled substance regulatory paths but only provides minor details on new pathways which are developed for upcoming therapeutic methods in Future Perspectives. The rapid pace of regulatory evolution requires organizations to update their guidance documents and fee schedules which will occur after the literature search deadline of March 2025. The narrative review format of this article introduces its basic research limitations because it uses narrative methods which create selection bias while failing to assess the research quality of its included studies.
The worldwide system for API approval regulations depends on the International Council for Harmonisation guidelines which began in 1990 as a joint project between European and Japanese and American regulatory bodies and pharmaceutical industry groups. The 2015 conversion of ICH into a legal organization enabled its membership to grow when it included new regulatory bodies and allowed CDSCO to join as an observer which helped to improve standardization. [3,17].
2.1 ICH Quality Guidelines for API Regulation
The ICH Quality (Q) guidelines establish a complete framework which governs all pharmaceutical quality requirements starting from API development and continuing through commercial production and post-marketing product management. The table 2 displays all essential ICH guidelines which apply to API regulations. The ICH Q7 standard exists within FDA regulations through its connection to 21 CFR Parts 210/211 interpretive guidance while it remains unrecognized as an independent federal regulation [18]. The production of API according to 21 CFR Parts 210/211 needs companies to follow ICH Q7 principles because they serve as necessary operational standards.
Table 1: ICH Guidelines for API Regulation — Comprehensive Summary
|
Guideline |
Full Title |
Key Content & Purpose |
Year |
Applicability |
Ref. |
|
ICH Q7 |
GMP for Active Pharmaceutical Ingredients |
Comprehensive GMP requirements for API manufacturing. Incorporated into FDA practice through 21 CFR 211 interpretive guidance; not independently codified as a federal regulation. |
2000 |
API manufacturers |
[18] |
|
ICH Q8(R2) |
Pharmaceutical Development |
Quality by design (QbD) principles, design space concept, risk-based pharmaceutical development, enhanced vs. minimal approaches. |
2009 |
Drug substance/product developers |
[19] |
|
ICH Q9(R1) |
Quality Risk Management |
Risk assessment, communication and review tools (FMEA, HACCP, FTA). Original Q9: 2005; (R1) revision adopted March 2023. |
2023 (R1) |
All pharmaceutical entities |
[17] |
|
ICH Q10 |
Pharmaceutical Quality System |
Lifecycle pharmaceutical quality system (PQS) encompassing development through discontinuation. |
2008 |
All pharmaceutical entities |
[20] |
|
ICH Q11 |
Development & Manufacture of Drug Substances |
API development philosophy, control strategy, process understanding, starting material selection and justification. |
2012 |
API manufacturers |
[21] |
|
ICH Q12 |
Pharmaceutical Lifecycle Management |
Post-Approval Change Management Protocol (PACMP), Established Conditions (EC), lifecycle change management tools. Implemented as FDA guidance 2023. |
2019 |
Manufacturers/regulators |
[22] |
|
ICH Q13 |
Continuous Manufacturing |
Regulatory framework for continuous manufacturing of APIs and drug products; batch definition, process validation for CM, RTRT. Final guideline 2023. |
2023 |
API/DP manufacturers |
[23] |
|
ICH Q14 |
Analytical Procedure Development |
Risk-based analytical procedure development, Method Operable Design Region (MODR). Reached Step 4 November 2023. |
2023 |
Analytical departments |
[24] |
|
ICH Q3A(R2) |
Impurities in New Drug Substances |
Organic impurity reporting, identification, and qualification thresholds for new APIs. |
2006 |
API manufacturers |
[25] |
|
ICH Q3C(R8) |
Residual Solvents |
Class 1/2/3 solvent classification, Permitted Daily Exposure (PDE) limits, risk-based approach to solvent control. |
2021 |
API manufacturers |
[26] |
|
ICH Q3D(R1) |
Elemental Impurities |
Risk assessment for 24 elements, PDE limits by route of administration. |
2019 |
API/DP manufacturers |
[27] |
|
ICH M7(R2) |
Assessment and Control of Mutagenic Impurities |
TTC approach; assessment and control strategy for DNA-reactive (mutagenic) impurities including nitrosamines. (R2) revision adopted March 2023. |
2023 (R2) |
API/DP manufacturers |
[28] |
|
ICH Q2(R2) |
Analytical Validation |
Validation characteristics, lifecycle approach to method validation, combined implementation with ICH Q14. |
2022 |
Analytical labs |
[29] |
|
ICH Q1A(R2) |
Stability Testing |
Stability study design, climatic zone conditions (Zone I–IVb), shelf-life determination for APIs and drug products. |
2003 |
API/DP manufacturers |
[30] |
|
ICH M4(R4) |
CTD Organisation |
Five-module Common Technical Document (CTD) structure for regulatory submissions; globally accepted by all ICH member authorities. |
2016 |
All applicants |
[3,4] |
The Common Technical Document (CTD) established by ICH M4 provides pharmaceutical companies with a standardized five-module framework which all ICH member regulatory bodies will accept for their regulatory submissions. [3,4]. The complete quality documentation of API DMF submissions exists as the primary quality dossier for Module 3 (Quality) and its Section 3.2.S (Drug Substance) component. Both CDSCO and USFDA base their CMC review on the CTD Module 3.2.S structure, though USFDA mandates strict eCTD electronic format while CDSCO recommends but does not yet mandate full eCTD [3,4].
2.3 eCTD Submission System
The electronic Common Technical Document (eCTD) standardizes regulatory submissions through a defined file folder structure, XML backbone, and electronic review capabilities. The USFDA required electronic Common Technical Document (eCTD) submission for all New Drug Applications, Abbreviated New Drug Applications, Biologics License Applications, and Investigational New Drug submissions starting from May 5 2017 for large companies and beginning on May 5 2018 for small companies that GDUFA defined according to the FDA Guidance for Industry on eCTD submissions which was published in January 2017 [29,20]. The FDA Electronic Submissions Gateway (ESG) serves as the platform for submission filing. The CDSCO in India advances its eCTD implementation through the SUGAM portal while the organization develops its mandatory implementation timeline which will take effect from March 2025 [13,16].
The API regulatory submission process across global regulatory systems follows a universal three-phase paradigm: (1) pre-submission preparation—compilation of manufacturing process data, analytical validation, impurity profiling, stability studies, process validation, GMP certification, and starting material justification; (2) formal submission—via eCTD/CTD-formatted DMF filing with regulatory fees; and (3) regulatory review—administrative completeness check, scientific CMC review, information request/query response cycles, GMP inspection, and approval/deficiency communication. The specific requirements, formats, timelines, and inspection standards within each phase differ substantially between CDSCO and USFDA, as detailed in Sections 3 and 4 [3,4,8].
India's pharmaceutical regulatory system functions as a vital component of the international drug supply chain which oversees more than 3000 licensed pharmaceutical manufacturers. [1,32] and a large number of USFDA-approved pharmaceutical facilities. The Central Drugs Standard Control Organisation (CDSCO), established under the Drugs and Cosmetics Act 1940 [10], The organization controls all aspects of pharmaceutical production activities which include manufacturing and distributing drugs and conducting quality assessments and API registration processes. CDSCO has developed into a more powerful organization through its various institutional upgrades which include improved technical abilities and the establishment of digital regulatory systems and its ongoing efforts to meet global regulatory standards. [10,11,32].
3.1 CDSCO Regulatory Structure
The CDSCO functions as a part of the Ministry of Health and Family Welfare (MoHFW) which the Drug Controller General of India (DCGI) leads according to his legal powers established by the D&C Act. The CDSCO Annual Report 2022-23 serves as the basis for this information. [32], the organisation operates a network which includes six zonal offices located in Mumbai Kolkata Chennai Hyderabad Guwahati and Ahmedabad together with four sub-zonal offices and thirteen port offices. The pharmaceutical regulations of India operate through a two-tier system which designates CDSCO as the central authority for new drug approvals and import licences while State Drug Authorities (SDAs) manage manufacturing licences and domestic GMP inspections. The bifurcation of the system creates operational difficulties because it leads to inconsistent enforcement procedures throughout different regions of India which contains 28 states and eight union territories. The revised Schedule M 2023 system presents The current regulation as a solution to this systemic issue. [11] partially addresses through more prescriptive national GMP standards.
The CDSCO-SDA coordination structure creates important practical effects which API manufacturers must address. A manufacturer may receive CDSCO approval for a new drug substance (via Form 33—the CDSCO application for new drug approval) yet experience delays in obtaining the SDA-issued manufacturing licence (Form 28D—a separate SDA-issued document) if the state authority has different inspection scheduling timelines or interpretation of GMP requirements. This dual-authority dynamic has no USFDA equivalent [10,32].
Organization Structure of CDSCO
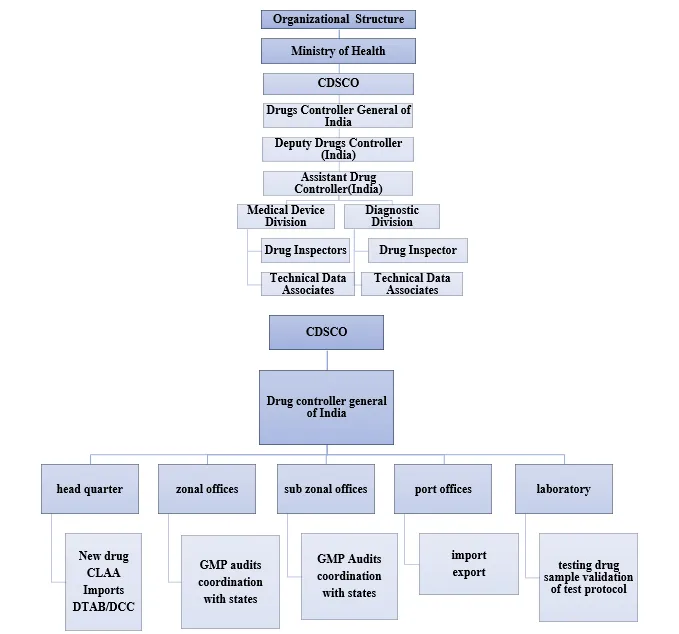
The Drugs and Cosmetics Act, 1940 (D&C Act) [10] The New Drugs and Clinical Trials Rules 2019 establish pharmaceutical regulations in India through their fundamental legal framework. The NDCT Rules 2019 serve as the primary legal framework that regulates pharmaceutical research activities. [33], The term 'new drug' will be defined to include any active pharmaceutical ingredient that has not received prior approval for use in India. The Schedule M section of the D&C Rules 2023 received extensive changes through the 28 March 2023 Gazette Notification G.S.R. 228(E) which introduced new regulations to the existing standards. [11], specifies GMP requirements for all pharmaceutical manufacturing including API production. The 2023 revision introduced enhanced requirements for data integrity (ALCOA principles), quality risk management (ICH Q9) [17], and pharmaceutical quality systems (ICH Q10) [20], substantially aligning India's GMP standards with international benchmarks for the first time [11,12,14].
3.3 Indian Drug Master File (IDMF)
The Indian Drug Master File (IDMF) is the primary regulatory dossier for API manufacturers supplying CDSCO-reviewed drug product applications. While conceptually modeled on the USFDA DMF system, the IDMF is less formally codified; CDSCO's 2018 Guidance Note on DMF [34] provides the primary reference document for IDMF preparation. The IDMF comprises a restricted part (confidential manufacturing process and quality data, accessible only to CDSCO) and an open part (administrative information available to drug product applicants). CDSCO recommends CTD Module 3.2.S format for IDMF organisation, facilitating alignment with international submission standards [3,4,34]. Unlike USFDA's formal five-type DMF classification [8], CDSCO does not publish a formal IDMF type classification, and there is no CDSCO equivalent of FDA's MAPP 3718.4 [40] procedural document for DMF review.
3.4 SUGAM Portal and Submission Process
The SUGAM portal (https://sugam.cdsco.gov.in) [13], The Central Drugs Standard Control Organization (CDSCO) uses its main digital regulatory system to handle all pharmaceutical applications. The system permits users to submit new drug applications and import license requests and manufacturing license requests and clinical trial applications online. The portal needs users to authenticate their identity through Digital Signature Certificate (DSC) and to upload electronic documents and to pay regulatory fees and to send electronic messages for CDSCO inquiries. SUGAM has made significant progress in improving regulatory efficiency, but the organization needs to implement complete eCTD according to ICH M4 specifications. [3,4] remains under development as of March 2025, with many submissions continuing in hybrid or primarily electronic-document formats [13,16].
3.5 Documentation Requirements
The documentation requirements for API registration in India are comprehensive because they demand complete data from all sections of CTD Module 3.2.S. The 2023 Schedule M revision [11] The organization established two new documentation categories which include explicit data integrity documentation through ALCOA compliance declaration and computerized system validation status and ICH Q11 requirements for enhanced starting material justification. [21] principles. Table 3 gives an extended review of all the issuance of permission, noticing, proof of evidence, and one could now see where the highly enforced categories rip in with. [11,33,34].
Table 2: Documents Required for API Registration in India (CDSCO)
|
Document/Data Element |
Specific Content Required |
CTD Module |
Status |
Ref. |
|
Application Form 33 |
Duly completed application for manufacture of new drug substance (NDCT Rules 2019); entity details; manufacturing site declaration; payment receipt. [Form 33 = CDSCO new drug application; Form 28D = separate SDA manufacturing licence] |
Module 1 |
Mandatory |
[33] |
|
Indian DMF (IDMF) |
Complete drug master file per CDSCO Guidance Note on DMF 2018; restricted part (confidential manufacturing and quality data) and open part (administrative reference information) |
Module 3.2.S |
Mandatory |
[34] |
|
Manufacturing Process (S.2.2) |
Full narrative description, reaction equations, flow diagram, in-process controls, critical steps, batch formula; starting material justification per ICH Q11 |
3.2.S.2 |
Mandatory |
[18,21] |
|
Process Validation (S.2.5) |
Prospective validation data for minimum three pilot/commercial batches; CQA/CPP identification; validation protocol and report per ICH Q7 |
3.2.S.2.5 |
Mandatory |
[18,46] |
|
Characterization (S.3.1) |
Elucidation of structure (IR, ¹H/¹³C NMR, MS, UV, X-ray crystallography); polymorphism characterisation; physicochemical properties |
3.2.S.3 |
Mandatory |
[18] |
|
Impurity Profile (S.3.2) |
Organic impurities (ICH Q3A) [25]; residual solvents (ICH Q3C) [26]; elemental impurities (ICH Q3D) [27]; genotoxic impurity fate/purge (ICH M7) [28]; nitrosamine risk assessment per CDSCO Circular 2022 [35] |
3.2.S.3.2 |
Mandatory |
[25–28,35] |
|
API Specifications (S.4.1) |
Justified specifications with all quality attributes, acceptance criteria, and compendial references (IP/BP/USP) |
3.2.S.4.1 |
Mandatory |
[18] |
|
Analytical Methods & Validation (S.4.2–4.3) |
Full description of all analytical procedures with method validation per ICH Q2(R2) [29]; system suitability criteria |
3.2.S.4.2–4.3 |
Mandatory |
[29] |
|
Stability Data (S.7) |
ICH Q1A Zone IVb: 40°C/75%RH long-term; 60°C/75%RH accelerated; minimum six months real-time data at submission; photostability per ICH Q1B |
3.2.S.7 |
Mandatory |
[30,48] |
|
Container Closure System (S.6) |
Details of primary packaging materials with specifications; extractables/leachables data; suitability for API protection |
3.2.S.6 |
Mandatory |
[11,33] |
|
GMP Certificate |
Valid WHO-GMP TRS 986 Annex 2 [28] or Schedule M 2023 [11] compliance certificate from competent State Drug Authority; current and covering the manufacturing site |
Module 1 |
Mandatory |
[11,28] |
|
Data Integrity Statement |
Declaration of ALCOA+ data integrity compliance; computerised system validation status; audit trail policy per revised Schedule M 2023 [11] |
Module 1 |
Mandatory (2023+) |
[7,12] |
|
Nitrosamine Risk Assessment |
Assessment per CDSCO Circular 2022 [35]; analytical confirmation (LC-MS/MS) if risk identified; proposed control strategy and AI limits |
3.2.S.3.2 |
Mandatory (2022+) |
[35,36,37] |
|
Starting Material Justification |
Justification for designation of drug substance starting materials per ICH Q11 principles [21]; lineage of synthesis from commercially available starting points |
3.2.S.2.3 |
Mandatory |
[21] |
3.6 Regulatory Timeline
The review timeline for API registration in India varies because it depends on three factors which include the complexity of the application and the completeness of the dossier and the duration of query cycles and inspection scheduling. Table 4 presents typical timelines for each process stage, derived from CDSCO Annual Report 2022–23 [32] and peer-reviewed literature [15,38]. The primary determinant of timeline variability is not CDSCO's scientific review capacity per se but rather the number of query cycles generated due to incomplete dossiers and SDA inspection scheduling delays [32,38].
Table 3: Regulatory Timeline for API Registration in India (CDSCO)
|
Stage |
Activity |
Approximate Duration |
Responsible Party |
Ref. |
|
Pre-Submission Preparation |
DMF compilation, analytical method development and validation, stability studies, process validation, GMP certification |
6–18 months |
Manufacturer |
[34,38] |
|
SUGAM Portal Registration & Submission |
Online application filing, dossier document upload (Form 33), regulatory fee payment, DSC authentication |
1–2 weeks |
Applicant |
[13,38] |
|
Administrative Completeness Check |
CDSCO screens submission for completeness; deficiency letter issued for incomplete applications within 30 days |
1–3 months |
CDSCO |
[33,34] |
|
Primary Technical Review |
Scientific evaluation by CDSCO technical officers; CMC review; expert queries generated |
3–8 months |
CDSCO |
[32,38] |
|
Expert Committee Review (NDAC) |
For complex or novel APIs: multi-expert evaluation by New Drugs Advisory Committee |
3–6 months (if triggered) |
CDSCO/NDAC |
[33,38] |
|
Query/Deficiency Letter Response |
Technical query communication to applicant via SUGAM; applicant responds with supporting data; multiple cycles possible |
1–6 months per cycle |
Applicant/CDSCO |
[34,38] |
|
GMP Inspection (CDSCO/SDA) |
On-site GMP inspection against Schedule M 2023 [11] and WHO-GMP TRS 986 Annex 2 [28]; CAPA required before approval; scheduling delays common |
1–3 months (scheduling variable) |
CDSCO/SDA |
[32,33] |
|
CAPA Verification |
Review and acceptance of Corrective and Preventive Action responses to inspection observations |
1–3 months |
CDSCO |
[33] |
|
Licence Grant (Form 28D) |
SDA manufacturing licence issuance; subject to annual renewal and ongoing GMP compliance |
2–4 weeks post-clearance |
CDSCO/SDA |
[33,34] |
|
Total Timeline — Straightforward Application |
For new API with complete dossier, one query cycle, and no major GMP observations |
12–24 months |
— |
[32,38] |
|
Total Timeline — Complex Application |
For novel API, multiple query cycles, or significant GMP deficiencies requiring remediation |
24–48+ months |
— |
[15,32] |
3.7 GMP Inspection Requirements
GMP inspection of API manufacturing facilities is a prerequisite for approval in India. CDSCO and SDAs conduct joint or separate inspections against Schedule M (revised 2023) [11] and WHO-GMP TRS 986 Annex 2 (2014) [28] requirements. Key evaluation areas include quality management systems, personnel qualifications, facility design and maintenance, equipment qualification, documentation integrity, production process controls, quality control laboratory operations, and complaint and recall management. The 2023 Schedule M revision introduced enhanced requirements for ALCOA-principal data integrity, quality risk management per ICH Q9 [17], and pharmaceutical quality systems per ICH Q10 [20].
India's admission to PIC/S as an observer in 2021 represents a watershed milestone in the international recognition of India's GMP inspection capabilities [14]. Full PIC/S membership, which the organization aims to obtain between 2025 and 2027, will provide CDSCO with the authority to negotiate MRAs that establish pharmaceutical inspection procedures with all other PIC/S full members through agreements that the US-EU MRA model. [31]— so that companies supplying various lucrative markets no longer need to comply with numerous second-party audits.
4. API Registration Process in the United States (USFDA)
The United States Food and Drug Administration (USFDA) maintain one of the world's most rigorous and scientifically sophisticated pharmaceutical regulatory systems. The USFDA establishes its regulations to control the pharmaceutical industry which operates as the largest market worldwide with a value of approximately USD 407 billion in 2022. [8], mandatory eCTD submission requirements [29,20], stringent DSSM designation standards [9], pre-approval inspection programs [42], and ALCOA+ data integrity enforcement [7]—are recognized as global regulatory benchmarks [5,6,8,39].
4.1 USFDA Regulatory Structure
The USFDA's regulatory structure for pharmaceuticals encompasses CDER (primary responsibility for new and generic drug review), the Office of Pharmaceutical Quality (OPQ) for pharmaceutical quality assessment, and the Office of Regulatory Affairs (ORA) for facility inspections. OPQ, which CDER established as a new office in 2015, combines CMC review from all application types (NDA, ANDA, BLA, IND) into one organization that provides consistent science-based evaluation of pharmaceutical quality. [39]. The existing structure of the integration system lacks a CDSCO equivalent because the organization distributes its CMC review process among multiple functional groups instead of using a specialized quality department.
4.2 Legal Framework
The Federal Food, Drug, and Cosmetic Act (FD&C Act) [5], The l duct sponsor. As of 2023, CDER manages more than 15,000 active DMFs globally [8,39,40], with Indian API manufacturers being the largest single country contributor.aw which came into effect in 1938 received major updates through the GDUFA III program which operates from fiscal year 2023 until fiscal year 2027. The essential regulations for API manufacturing operations require compliance with 21 CFR Part 210 which establishes basic GMP requirements and 21 CFR Part 211 which defines GMP standards for complete pharmaceutical products. [6], The regulation 21 CFR Part 11 establishes requirements for electronic records and electronic signatures. The 21 CFR Part 284 regulation establishes rules for drug product applications which include New Drug Applications and Abbreviated New Drug Applications while API manufacturers must follow 21 CFR Parts 210 and 211. [6]. ICH Q7 [18], The document functions as FDA interpretive guidance which establishes specific Good Manufacturing Practice requirements that apply to Active Pharmaceutical Ingredient production according to 21 CFR Parts 210 and 211.
4.3 Drug Master File (DMF) System
USFDA DMF system is the most sophisticated and systemic DMF system in the world, with clear procedural needs, as outlined in FDA MAPP 3718.4 for visitors interested in the DMF process. [40] and the 2018 DMF Guidance for Industry [8]. A Type II DMF—the predominant DMF type for API manufacturers—is a voluntary submission of confidential manufacturing information to FDA that authorizes specific drug product applicants to reference the DMF in ANDA, NDA, or IND applications without disclosing proprietary manufacturing details to the drug product.
Table 4: Drug Master File (DMF) Types in the USA — Classification and Application
|
DMF Type |
Category |
Specific Content Covered |
Key Users |
Ref. |
|
Type I |
Manufacturing Site, Facilities, Operating Procedures and Personnel |
Manufacturing site information, facility layout, organization charts, SOPs, personnel qualifications. Rarely filed independently; most site information incorporated into Type II. |
Contract manufacturers, multi-product sites |
[8,40] |
|
Type II |
Drug Substance, Drug Substance Intermediate, and Material Used in Their Preparation; Drug Product |
PRIMARY DMF FOR APIs: Full CTD Module 3.2.S data including manufacturing process, characterization, impurity controls, stability, process validation; also covers intermediates and excipients manufactured by drug substance manufacturers. |
API manufacturers (most common DMF type) |
[8,18,40] |
|
Type III |
Packaging Material |
Container closure systems; primary/secondary packaging materials; specifications; extractables and leachables data; suitability for API packaging. |
Packaging material suppliers, glass/plastic manufacturers |
[8,40,54] |
|
Type IV |
Excipient, Colorant, Flavor, Essence, or Material Used in Their Preparation |
Inactive pharmaceutical ingredients: identity, purity, specifications, manufacturing process; European Certificate of Suitability (CEP) may substitute for some excipients. |
Excipient manufacturers, food additive suppliers |
[8,40] |
|
Type V |
FDA Accepted Reference Information |
Miscellaneous technical information not covered under Types I–IV; includes master files for novel synthesis platforms or process analytical technology (PAT) platforms. |
Various; case-by-case assessment |
[8,40] |
4.4 Drug Substance Starting Material (DSSM) Designation — A Critical USFDA Requirement
One of the most practically significant and frequently misunderstood requirements for USFDA Type II DMF submissions is the proper designation of drug substance starting materials (DSSMs) per FDA's Guidance for Industry: Drug Substance Starting Material Designation in ANDAs and NDAs (2022) [9]. The DSSM Guidance 2022 of the FDA establishes formal regulations for ANDAs and NDAs while the DSSM designation assessment process impacts Type II DMF content through Information Requests which result from the application review process. FDA applies the principles of ICH Q11 [21] to require that starting materials be: (1) commercially available from multiple sources, (2) adequately characterized chemical entities, and (3) at a point in the synthetic route where the synthesis does not introduce impurities of concern (including genotoxic impurities per ICH M7 [28]) that cannot be adequately controlled in the subsequent steps [9,21].
DSSM designation serves essential functions for Indian API manufacturers which cannot be diminished. The FDA Information Requests (IRs) which dispute starting material designation represent one of the most commonly reported deficiency types in Type II DMF assessments conducted by Indian API manufacturers. The use of early-route chemical starting materials by manufacturers results in DSSM-related IRs which lead to significant delays in review processes. [9,21]. CDSCO has not issued any guidance that is equivalent to the DSSM and this results in a sizeable practical difference from the USFDA requirements.
4.5 DMF Submission Process
Pre-submission regulatory strategy and data compilation; (2) mandatory eCTD format assembly per ICH M4 [3,4] and FDA eCTD specifications [29,20]; (3) electronic submission via FDA ESG with DMF cover sheet (FDA Form 3794) and GDUFA III fee payment [41]; (4) FDA acknowledgement letter with assigned DMF number within 30 calendar days; (5) administrative completeness review (Refuse-to-Review assessment within 60 days); (6) full technical review by the OPQ CMC team; (7) Information Request (IR) or deficiency letter issuance; (8) Pre-Approval Inspection of API manufacturing facility [42]; (9) DMF Satisfactory Determination.
4.6 Documentation Requirements
The documentation requirements when submitting Type II DMF to USFDA are comprehensive and must be arranged in a structured eCTD format that corresponds to CTD Module 3.2.S.Table 6 provides a detailed overview of all required data elements, organized by CTD section.
Table 5: USFDA Type II DMF Documentation Requirements by CTD Section
|
CTD Section |
Specific Content Required |
Guidance Reference |
Criticality |
Ref. |
|
Module 1 (Regional) |
Form 206h; GDUFA III DMF user fee payment evidence [41]; labeling; patent certifications (Para I–IV for ANDA); debarment certification; field copy certification |
21 CFR Parts 210, 211 for GMP; GDUFA III [41] |
Mandatory |
[5,6,41] |
|
3.2.S.1 General Info |
Nomenclature (INN, CAS, USAN); structural formula; molecular formula/weight; general properties (pKa, aqueous solubility, partition coefficient, polymorphic forms, chirality) |
ICH Q6A [56], USP <1237> |
Mandatory |
[56] |
|
3.2.S.2 Manufacture |
S.2.1: Manufacturer(s) and sites; S.2.2: Description of manufacturing process; S.2.3: Control of materials including starting material justification per FDA DSSM guidance [9]; S.2.4: Controls of critical steps and intermediates; S.2.5: Process validation/evaluation [46]; S.2.6: Manufacturing process development history |
ICH Q7 [18], Q11 [21], Q8(R2) [19]; FDA DSSM Guidance [9]; FDA PV Guidance [46] |
Mandatory |
[9,18,19,21,46] |
|
3.2.S.3 Characterization |
S.3.1: Elucidation of structure (full spectroscopic characterization: ¹H/¹³C NMR, IR, MS, UV, X-ray crystallography); polymorph characterization; S.3.2: Impurity profile — organic impurities (ICH Q3A) [25], residual solvents (ICH Q3C) [26], elemental impurities (ICH Q3D) [27], genotoxic impurity fate and purge assessment (ICH M7(R2)) [28], nitrosamine risk assessment and control strategy [36] |
ICH Q3A [25], Q3C [26], Q3D [27], M7(R2) [28]; FDA 2021 Nitrosamine Guidance [36] |
Mandatory |
[25–28,36] |
|
3.2.S.4 Controls |
S.4.1: Specification with acceptance criteria justified against batch data and stability; S.4.2: Analytical procedures (full description); S.4.3: Validation of analytical procedures (ICH Q2(R2)) [29]; S.4.4: Batch analyses (CoA ≥3 batches); S.4.5: Characterization of impurities; S.4.6: Justification of specification |
ICH Q2(R2) [29], Q6A [56], USP |
Mandatory |
[29,56] |
|
3.2.S.5 Reference Standards |
Primary reference standard characterization or compendial reference (USP/EP/BP); secondary/working standard qualification protocol; certificate of analysis |
USP, ICH Q6A [56] |
Mandatory |
[56] |
|
3.2.S.6 Container Closure |
Container closure system description and specifications; suitability data (protective properties, compatibility, safety); extractables/leachables data per USP <447>, <1663>, and <1646> |
FDA Container Closure Guidance 1999 [54]; USP <447>, <1663>, <1646> |
Mandatory |
[54] |
|
3.2.S.7 Stability |
S.7.1: Stability summary and conclusions; S.7.2: Post-approval stability protocol and commitment; S.7.3: Stability data (ICH Q1A Zone II: 25°C/60%RH long-term minimum 12 months at submission; 40°C/75%RH accelerated 6 months; photostability per ICH Q1B) |
ICH Q1A(R2) [30], Q1B, Q1E |
Mandatory |
[30] |
|
Starting Material Designation Justification |
Written justification for selection of drug substance starting material (DSSM) designation; demonstration that starting material is commercially available, adequately characterised, and synthesis route does not introduce unacceptable impurities; response to FDA DSSM guidance |
FDA DSSM Guidance 2022 [9] |
Mandatory |
[9,21] |
|
Nitrosamine Risk Assessment |
Documented risk assessment for all potential nitrosamine formation pathways; confirmatory LC-MS/MS analytical testing if risk identified; proposed control strategy and limits per FDA 2021 guidance [36] |
FDA 2021 Nitrosamine Guidance [36] |
Mandatory (2021+) |
[36,37] |
|
Data Integrity Declaration |
Confirmation that all data submitted comply with 21 CFR Part 11 (electronic records and signatures) and ALCOA+ data integrity principles |
21 CFR Part 11; FDA DI Guidance 2018 [7] |
Mandatory |
[7,12] |
4.7 GMP Inspection and Pre-Approval Inspection (PAI)
USFDA's pre-approval inspection (PAI) programme represents the most comprehensive API facility evaluation system globally. PAIs are conducted by ORA field inspectors and are triggered by receipt of a DMF-referencing ANDA or NDA application. USFDA's site selection model for PAIs incorporates risk-based prioritization factors including facility compliance history, time since last inspection, product risk profile (sterile vs. non-sterile, controlled substance), throughput volume, and geographic region [42].
Data integrity enforcement has emerged as the defining challenge in USFDA's inspection programme for Indian API manufacturers. Analysis of USFDA enforcement actions [43,44] identified data integrity violations—including audit trail manipulation, backdated records, selective data reporting, and unauthorized data deletion—as the most prevalent observation category in Warning Letters issued to Indian pharmaceutical facilities between 2014 and 2022. USFDA's 2018 Data Integrity Guidance [7] The organization needs to follow ALCOA+ standards for all GMP records and requires validated audit trails for all computerized systems. The trend data indicates that data integrity-related Warning Letters to Indian facilities have decreased since 2019 because facilities started making systematic investments in their quality systems. [43,44].
The study uses a comparative analysis to examine how API registration regulations in India and the United States. The study demonstrates that both countries share ICH foundations which create a path for their regulatory frameworks to achieve complete commonality yet their actual implementation practices show different levels of development which affects their digital systems and regulatory enforcement methods and their system of official regulations and their particular technical requirements
Table 7 presents a comprehensive assessment which compares CDSCO and USFDA regulatory systems through 20 essential aspects of API registration. The table includes current GDUFA III fee information for fiscal year 2024 [41] The three parameters which previous comparative studies have not addressed include starting material designation standards and CEP recognition policy and post-approval change framework comparison.
Table 6: Comprehensive Comparison of India (CDSCO) vs. USA (USFDA) API Regulatory Framework
|
Parameter |
India (CDSCO) |
USA (USFDA) , REF |
|
Primary Regulatory Authority |
CDSCO under Ministry of Health and Family Welfare; headed by DCGI [10,11] |
USFDA — CDER; Office of Pharmaceutical Quality (OPQ) established 2015 for integrated CMC review [5,6,39] |
|
Jurisdiction Structure |
Two-tier: Central (CDSCO) for new drug approval and import licences + State Drug Authorities (SDAs) for manufacturing licences and domestic GMP inspections [10,32] |
Exclusive unified federal authority: CDER/OPQ for scientific review; ORA for facility inspections [5,6,8] |
|
Primary Governing Legislation |
Drugs and Cosmetics Act, 1940; D&C Rules 1934; New Drugs and Clinical Trials Rules, 2019 [33,34] |
Federal Food, Drug, and Cosmetic Act (FD&C Act); 21 CFR Parts 210, 211; GDUFA III (FY2023–2027) [5,6,41] |
|
GMP Standards |
Schedule M (revised 2023, G.S.R. 228(E)) [11] aligning with WHO-GMP TRS 986 Annex 2 [28] and ICH Q7 principles [18]; ALCOA data integrity principles introduced; PIC/S observer (2021) [14] |
21 CFR Parts 210/211 [6]; ICH Q7 [18] incorporated as FDA interpretive guidance (not an independent federal regulation); ALCOA+ strictly enforced; robust Warning Letter enforcement history [5,7,12,18] |
|
DMF System |
Indian Drug Master File (IDMF): less formally codified; governed by CDSCO Guidance Note 2018 [34]; restricted part and open part; progressively aligned with CTD Module 3.2.S format [34,60] |
Formal five-type DMF (Types I–V) [8,40]; Type II for APIs; detailed procedural requirements in FDA MAPP 3718.4 [40] and DMF Guidance 2018 [8]; >15,000 active DMFs managed by CDER [8,39,40] |
|
Submission Format |
SUGAM portal [13] (hybrid digital-paper); full eCTD submission not yet mandatory; eCTD adoption planned [13,16] |
Mandatory eCTD (XML backbone) via FDA Electronic Submissions Gateway (ESG) since May 5, 2017 (large companies) and May 5, 2018 (small companies) per FDA eCTD Guidance 2017 [29,20] |
|
CTD Compliance |
CTD Module 3.2.S format recommended but not strictly mandated; progressive adoption encouraged [3,4,34] |
CTD/eCTD mandatory for all NDA, ANDA, BLA, and IND submissions; full ICH M4 compliance required [3,4,29] |
|
Starting Material Designation |
ICH Q11 [21] principles recommended but FDA-equivalent DSSM-specific guidance not yet published by CDSCO |
Stringent designation criteria per FDA DSSM Guidance 2022 [9]; starting materials must be commercially available, adequately characterized, and synthesis routes must not introduce impurities of concern. Note: DSSM Guidance formally addresses ANDAs and NDAs; DMFs are affected through referencing application review [9,21] |
|
User Fees |
Nominal regulatory fees; SUGAM portal processing fees; no facility-level annual fee equivalent to GDUFA [33,34] |
Substantial GDUFA III fees (FY2024) [41]: Type II DMF fee ~USD 62,420; facility fee ~USD 388,137; ANDA filing fee ~USD 109,993. Annual fees indexed to inflation. Fee waivers available for qualifying small businesses [41,45] |
|
GMP Inspection Type |
CDSCO and/or SDA joint or separate domestic inspections; WHO-GMP inspections for export certification; India PIC/S observer since 2021 [11,14] |
Pre-Approval Inspection (PAI) mandatory for new API sources; unannounced surveillance inspections; risk-based site selection model [42]; ORA international inspection offices worldwide [8,39] |
|
Impurity Requirements |
ICH Q3A [25], Q3C [26], Q3D [27] adopted; CDSCO 2022 Circular [35] on nitrosamine risk assessment; ICH M7 increasingly aligned |
ICH Q3A [25], Q3C [26], Q3D [27], M7(R2) [28] all enforced; FDA 2021 Nitrosamine Guidance [36] mandatory; genotoxic impurity fate and purge assessment required; ultra-sensitive LC-MS/MS analytical methods required |
|
Data Integrity Framework |
ALCOA principles adopted in revised Schedule M 2023 [11]; CDSCO Data Integrity Guidelines 2022 [12] aligned with MHRA/WHO guidance [80]; enforcement increasing |
ALCOA+ principles strictly enforced [7]; 21 CFR Part 11 for electronic records and signatures; FDA Data Integrity Guidance 2018 [7]; data integrity citations most frequent in Warning Letters to Indian facilities 2014–2022 [43,44] |
|
Post-Approval Changes |
Variation applications for major post-approval changes; CBE notification for minor changes; India-specific SUPAC-equivalent guidance absent—creates filing category ambiguity [33,62] |
SUPAC guidelines for scale-up and post-approval changes; CBE-0, CBE-30, Prior Approval supplement categories clearly defined by change type; ICH Q12 [22] PACMP implemented as FDA guidance 2023 [22,63] |
|
Stability Requirements |
ICH Q1A Zone IVb (40°C/75%RH) long-term; 60°C/75%RH accelerated; minimum 6 months real-time data at submission for new applications [30,48] |
ICH Q1A Zone II (25°C/60%RH) long-term; 40°C/75%RH accelerated; minimum 12 months real-time data at ANDA submission [30] |
|
CEP Recognition |
CDSCO increasingly considers EDQM CEPs as partial quality evidence; policy evolving without published formal guidance. No specific CDSCO policy document available. |
FDA does not formally accept EDQM CEPs in lieu of DMF data; separate Type II DMF required for all API sources referenced in US applications [8,40] |
|
Regulatory Transparency |
Moderate; SUGAM portal [13] improves submission tracking; approval letters and detailed review documents not routinely publicly available |
High: Drugs@FDA public database; FDA approval letters and review documents published; Warning Letters publicly searchable; FDA FACTS inspection database [5,39] |
|
International Recognition |
PIC/S observer (2021) [14]; WHO-GMP certificates widely accepted in developing markets; CDSCO working toward PIC/S full membership and potential MRA negotiations [14,31] |
Global regulatory benchmark; ICH founding member; US-EU MRA fully implemented 2019 [31]; bilateral inspection cooperation with 30+ countries [14,31] |
|
Pharmacovigilance |
Post-marketing surveillance through ADR reporting system; Pharmacovigilance Programme of India (PVPI); API manufacturers responsible for safety reporting [33] |
MEDWatch adverse event reporting; post-marketing commitment (PMC) studies; FDA safety communications; Risk Evaluation and Mitigation Strategies (REMS) for high-risk drugs [5] |
5.2 Regulatory Authority Structure: Analytical Evaluation
The most significant structural difference between CDSCO and USFDA is the two-tier system between the center and the states. In other words, an API manufacturer in India has to comply with two different regulations: one from the center (CDSCO for new drug approval through Form 33) and the other from the state (SDA for the manufacturing license through Form 28D), for the same API at the same manufacturing site [10, 32]. In the case of the USFDA, the single system at the federal level means that one body regulates both the application for approval (CDER/OPQ and the manufacturing site (ORA), with the entire process being handled through the FDA system [8, 39].
State-level enforcement variability has also been proposed in the regulatory reform literature as having historically provided an opportunity for inconsistent compliance outcomes, whereby some manufacturers may experience less stringent inspections compared with others [15, 38, 46]. The 2023 Schedule M revision [11]—in that it provides a more prescriptive, uniform GMP standard across the nation—and PIC/S observer engagement [14]—in that it provides international peer review for the quality of an inspection program—both serve as a countermeasure for state-level variability.
5.3 Starting Material Designation: A Critical Divergence
The difference between the USFDA (official Guidance 2022 [9]) and CDSCO (ICH Q11-recommended principles, without any published guidance) in the DSSM designation is arguably one of the most critical differences for an Indian API manufacturer. An Indian API manufacturer may prepare a CTD Module 3.2.S dossier, which is meant for CDSCO, and designate a relatively early route starting material, thereby minimizing the scope of synthetic chemistry data included in the submission. However, if this same dossier is referenced in a USFDA Type II DMF, an IR will likely be issued challenging the starting material designation, which will necessitate either (a) a redesignation of a later, more complex starting material and a greatly expanded CTD data package, or (b) an amended synthetic route that qualifies the current starting material against FDA's criteria [9, 21]. Indian API manufacturers seeking CDSCO and USFDA registration for the API will need to design a starting material strategy that meets the criteria for the FDA's DSSM Guidance [9] from the outset, even if CDSCO will accept an earlier route starting material. ICH Q11 [21] principles are available for this purpose, even in the absence of CDSCO-specific DSSM Guidance.
5.4 Submission Format and Digital Infrastructure
USFDA's mandatory eCTD requirement (operational since 2017–2018) [29,20] represents the most practically impactful difference in submission infrastructure. eCTD's XML backbone, structured folder organization, electronic hyperlinks, and automated validation capabilities enable faster processing, electronic query management, and comprehensive review tracking. India's SUGAM portal [13], Although representing significant progress, this does not yet support the submission of full eCTDs, and the move towards mandatory eCTDs for CDSCO, which is currently in planning, will require significant investment in eCTD authoring skills and document management systems on the part of Indian API manufacturers [13,16].
5.5 Approval Timeline Comparison — Analytical Assessment
A revised approval timeline comparison chart is given below in Table 8, where the previously used estimated timelines have been replaced by actual timelines based on the CDSCO Annual Report 2022-23 [32] and performance reports of GDUFA III [45]. The most important difference while analyzing is the fact that the USFDA ANDA review timeline (12 months as per GDUFA III performance goal [41]) and PAI completion timeline (6 to 18 months post PAI assignment [42]) run parallel and not sequentially. The DMF does not have a separate ‘approval’ but rather a Satisfactoriness Determination as part of reference [8,41,45].
Table 7: Approval Timeline Comparison — India (CDSCO) vs. USA (USFDA)
|
Stage |
India Duration |
USA Duration |
India Effort |
USA Effort |
References |
|
Pre-Submission Preparation |
6–18 months |
3–12 months |
High |
High |
[8,34,38] |
|
Administrative Review / Completeness Check |
1–3 months |
1–2 months (RTR within 60 days) |
Low |
Low |
[8,33,45] |
|
Primary Technical Review (CMC) |
3–8 months |
Per GDUFA III [41]: standard ANDA goal 12 months total |
Medium |
High |
[32,41,45] |
|
Expert Committee / Pre-Approval Inspection (PAI)† |
3–6 months (NDAC, if triggered) |
6–18 months (PAI scheduling and conduct; concurrent with ANDA review) |
Medium |
Very High |
[8,33,42] |
|
Query/Information Request Response Cycle(s) |
1–6 months per cycle (multiple cycles common) |
2–6 months per CRL/IR cycle |
High |
High |
[32,38,45] |
|
GMP Inspection & CAPA Verification |
1–3 months (inspection) + 1–3 months (CAPA) |
6–18 months (PAI scheduling; international inspections prioritised by risk) |
High |
Very High |
[8,32,42] |
|
Licence/Approval Grant |
2–4 weeks post clearance |
1–3 months post PAI satisfactoriness |
Low |
Low |
[8,33] |
|
TOTAL — Typical (Straightforward Application) |
12–24 months |
18–30 months (ANDA approval including PAI) |
— |
— |
[8,32,38,45] |
|
TOTAL — Complex Application |
24–48+ months |
24–48+ months |
— |
— |
[8,32] |
5.6 Post-Approval Changes: Regulatory Framework Comparison
Both regulatory systems maintain frameworks for managing post-approval changes to approved API registrations, but differ substantially in specificity and ICH Q12 [22] implementation status. Table 13 provides the first dedicated comparison of post-approval change frameworks between CDSCO and USFDA in this article.
Table 8: Post-Approval Change Management — India (CDSCO) vs. USA (USFDA)
|
Parameter |
India (CDSCO) |
USA (USFDA), Ref |
|
Governing Framework |
Variation application system under NDCT Rules 2019 [33]; CBE notification for minor changes; no India-specific SUPAC-equivalent guidance published; ambiguity in change classification creates potential for under- or over-reporting |
SUPAC guidelines (product-category specific—SUPAC-IR [63] applies to solid oral dosage forms, not directly to APIs); CBE-0 (annual report), CBE-30 (prior notification), and Prior Approval supplement categories clearly defined by change type and potential quality impact [63] |
|
ICH Q12 Implementation |
CDSCO alignment with ICH Q12 [22] PACMP and Established Conditions anticipated; not yet formally implemented as CDSCO guidance |
Implemented as FDA guidance (January 2023); PACMP enables manufacturers to pre-agree post-approval change pathways with FDA, reducing approval timelines for pre-defined changes [22] |
|
Established Conditions (EC) |
Concept not yet formally adopted by CDSCO; manufacturers recommended to align with ICH Q12 principles proactively |
Formally implemented: Established Conditions (ECs) define approved manufacturing parameters that require Prior Approval if changed; differentiated from non-ECs that require CBE-30 or CBE-0 only [22] |
|
Post-Approval Stability Commitment |
Ongoing stability studies required per initial submission commitment; annual report submission |
Annual Report filing required within 60 days of DMF anniversary date; ongoing stability data must be submitted; failure to file annual report can result in DMF closure [8] |
|
Manufacturing Site Change |
Variation application to CDSCO; may require inspection of new site; timeline 6–18 months |
Prior Approval supplement required; PAI of new manufacturing site mandatory before approval; SUPAC MaSC guidance (API manufacturing site change—separate from SUPAC-IR) defines documentation requirements [63] |
|
Starting Material Change |
Variation application if starting material designation changes; CDSCO assessment |
Prior Approval supplement required if change involves starting material designation; FDA DSSM guidance 2022 [9] applies; significant scrutiny for changes that alter impurity profile [9,63] |
The registration of APIs in controlled pharmaceutical markets creates multiple difficulties which include regulatory and scientific and operational and financial aspects. Indian API manufacturers face additional challenges because they must handle two main regulatory systems while their technical requirements increase and regulatory changes for nitrosamine impurities and data integrity and DSSM designation and digital submissions evolve at a fast rate. [15,46].
6.1 Regulatory Challenges and Evidence-Grounded Solutions
Table 9 provides a systematic overview of the principal regulatory challenges, incorporating two new challenge categories not in prior literature: DSSM designation challenges specific to USFDA, and post-approval change management ambiguity.
Table 9: Regulatory Challenges in API Registration — India vs. USA, with Recommended Strategies
|
Challenge Category |
Specific Challenge |
Recommended Strategies and Solutions |
Ref. |
|
Regulatory Complexity |
Simultaneous navigation of CDSCO and USFDA requirements with divergent documentation formats; inconsistent application of standards between central and state authorities in India |
Dedicated regulatory affairs teams for each jurisdiction; regulatory intelligence services; harmonized CTD-format dossier preparation as common base for all markets |
[15,46,47] |
|
Documentation Burden |
Comprehensive CTD Module 3.2.S dossiers with stability studies requiring 12–24 months; process validation across multiple commercial-scale batches; starting material justification |
Early regulatory planning integrated with API development phase; modular dossier preparation; electronic document management systems (EDMS); parallel preparation of CDSCO and USFDA dossiers |
[3,4,34] |
|
GMP Compliance — Data Integrity |
Data integrity violations most frequent citation in USFDA Warning Letters to Indian facilities (2014–2022) [43,44]; ALCOA+ implementation challenges in legacy computerised systems; audit trail integrity |
Validated LIMS and ERP systems with 21 CFR Part 11 [6]-compliant audit trails; data integrity training programmes; management commitment to quality culture; third-party data integrity audits |
[7,12,43,44] |
|
GMP Compliance — Schedule M and 21 CFR |
Gap analysis required against revised Schedule M 2023 [11]; equipment re-qualification; process re-validation; enhanced QRM and data integrity documentation requirements |
Systematic gap analysis against Schedule M 2023 [11] and ICH Q7 [18]; risk-based remediation planning; capital investment in facility upgrades; QMS implementation per ICH Q10 [20] |
[11,14,18,28] |
|
Starting Material Designation (USFDA-specific) |
FDA DSSM Guidance 2022 [9] imposes stringent criteria not yet matched by CDSCO; Information Requests (IRs) for inadequate justification are a leading cause of API DMF deficiencies |
Early engagement with FDA DSSM guidance [9]; ICH Q11 [21]-aligned starting material selection during development; pre-submission meetings with FDA to align designation; synthetic route design to qualify appropriate starting points |
[9,21] |
|
Inspection Readiness |
USFDA PAI preparation for international inspections; unpredictable scheduling; documentation system organisation; potential unannounced inspections after initial approval |
Continuous inspection readiness programmes; pre-PAI readiness assessments; mock inspections by qualified consultants; robust CAPA management systems; front room/back room protocols |
[8,42,66] |
|
Nitrosamine Impurity Assessment |
Complex multi-step synthesis risk assessment across all reagents, solvents, and degradation pathways; ultra-sensitive LC-MS/MS analytical method development/validation; potential need for process redesign |
Early nitrosamine risk assessment during development; proactive analytical capability investment (LC-MS/MS); process risk mitigation at design stage; engagement with specialized analytical CROs; regulatory submissions per FDA 2021 guidance [36] and CDSCO 2022 circular [35] |
[36,37,67,68] |
|
eCTD Transition Costs (India) |
Investment in eCTD authoring software, XML backbone expertise, document management infrastructure, and submission validation before CDSCO mandates transition |
eCTD authoring tool selection and implementation; regulatory publishing team training; phased implementation; outsourcing to specialist regulatory publishing CROs for initial eCTD submissions |
[13,16] |
|
User Fee Burden (USFDA) |
GDUFA III [41] facility fees, DMF fees, and ANDA filing fees constitute substantial compliance costs; proportionally higher burden for small and medium manufacturers |
GDUFA fee waiver applications for qualifying small businesses; strategic DMF filing to cover multiple drug products; government PLI scheme support [69]; consortium compliance platforms for smaller manufacturers |
[41,45,69] |
|
Post-Approval Change Management |
Divergent change classification frameworks between India and USA; India lacks SUPAC-equivalent specific guidance; risk of out-of-cycle manufacturing changes requiring regulatory approval |
ICH Q12 [22] lifecycle management planning; PACMP development for pre-agreed change pathways; change control systems linked to regulatory change classification SOPs; India-specific variation guidance development |
[22,62,63] |
International regulatory harmonization represents the most powerful and sustained force reshaping the global pharmaceutical regulatory landscape. ICH guidelines, now adopted by regulatory authorities in more than 20 countries and regions, have substantially reduced the regulatory fragmentation that previously required pharmaceutical manufacturers to prepare fundamentally different dossiers for each major regulatory jurisdiction [3,17,23]. A nuanced analytical point is that harmonization also concentrates regulatory risk: the 2018 nitrosamine impurity crisis—which occurred despite APIs passing all applicable ICH-aligned specifications—illustrates this concentration risk [36,37]. Regulatory harmonization must therefore be accompanied by robust post-marketing surveillance systems.
7.1 Global Regulatory Harmonization Initiatives
Table 10 provides an updated overview of major global regulatory harmonization initiatives relevant to API registration, incorporating important clarifications about the US-EU MRA implementation timeline [31] and updated PIC/S membership count [14].
Table 10: Global Regulatory Harmonization Initiatives and Their Impact on API Registration
|
Initiative |
Governing Body |
Key Elements and Impact |
Relevance to India–USA |
References |
|
ICH Quality Guidelines (Q7–Q14) |
ICH (industry + regulators) |
Q7–Q14: API GMP, QbD, risk management, quality systems, lifecycle management, continuous manufacturing, analytical development. Progressively adopted by CDSCO and USFDA as mandatory or guidance-level requirements. |
Primary harmonization framework for both jurisdictions; CDSCO Schedule M 2023 revision substantially ICH Q7-aligned |
[17–24] |
|
WHO Prequalification Programme |
WHO |
Quality certification for medicines supplied to UN procurement agencies; GMP assessment of API and FPP manufacturers globally; India holds largest number of WHO-prequalified sites globally. |
India's WHO-PQ site leadership reflects commitment to international quality; pathway for developing-market access beyond India/USA |
[28–33] |
|
PIC/S GMP Scheme |
PIC/S (41+ participating authorities as of 2024–2025; verify current count at picscheme.org) |
Harmonized GMP inspection standards; mutual exchange of GMP inspection reports; inspector training and benchmarking. India admitted as observer 2021 [14]; progress toward full membership ongoing. |
India as PIC/S observer (2021); pathway toward full membership and inspection equivalence, potentially enabling MRA negotiations with USFDA |
[14,31] |
|
US-EU Mutual Recognition Agreement (MRA) |
FDA and EU competent authorities |
Pharmaceutical annexe signed 1998; re-activated November 2017 for initial EU member states; full implementation for all 27 EU member states achieved March 2019. Model for future India-USA inspection cooperation. |
Template for potential India-USA inspection information sharing post PIC/S full membership; reduces duplicative inspections and costs |
[31,42] |
|
ICH M4 CTD/eCTD |
ICH |
Standardized five-module CTD structure; eCTD electronic format mandated by USFDA/EMA/PMDA/Health Canada; CDSCO eCTD adoption planned. Enables single dossier adaptable across jurisdictions with Module 1 regional modifications only. |
Foundation for harmonized submissions; reduces multi-market preparation burden; India-USFDA convergence accelerating |
[3,4,29,20] |
|
ICH Q12 Lifecycle Management |
ICH |
PACMP and Established Conditions enable pre-agreed post-approval change pathways; implemented as FDA guidance 2023; CDSCO alignment anticipated. |
Reduces post-approval change delays; Indian manufacturers can leverage PACMP to pre-negotiate change protocols with FDA and eventually CDSCO |
[22,62,63] |
|
ICH Q13 Continuous Manufacturing |
ICH |
Regulatory framework for continuous manufacturing of APIs and drug products; batch definition for CM; real-time release testing (RTRT) framework. Final guideline 2023. |
Both CDSCO and USFDA expected to update guidance per ICH Q13 2023; growing number of CM-API approvals globally |
[23,70] |
|
ALCOA+ Data Integrity Standards |
USFDA, MHRA, EMA, WHO, CDSCO |
Global convergence on ALCOA+ (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available) data integrity principles across all regulatory systems. CDSCO Data Integrity Guidelines 2022 [12]. |
CDSCO Schedule M 2023 [11] incorporates ALCOA principles; convergence with USFDA standard narrowing enforcement gap |
[7,12,14] |
Significantly, the API registration regulations have a profound and multi-dimensional impact on the API businesses. In this regard, the API registration regulations impact the API businesses in terms of market access, investment strategies, competitive positioning, the structure of the API businesses' chain, and finally the availability of affordable medicines to the end-users. In this regard, the API businesses in India, the largest pool of generic API manufacturers globally, face both challenges and opportunities by virtue of being compliant with both CDSCO and USFDA regulations. [1,2,48].
8.1 Regulatory Impact on the API Pharmaceutical Industry
Table 11: Impact of Regulatory Frameworks on the API Pharmaceutical Industry
|
Impact Domain |
Specific Impact on API Industry |
Strategic Implications |
References |
|
Market Access — India |
CDSCO approval required for domestic API sales and for Certificate of Pharmaceutical Product (CoPP) issuance; Schedule M 2023 [11] compliance mandatory for all licensed manufacturing sites; WHO-GMP TRS 986 Annex 2 [28] required for regulated-market export |
Investment in dual regulatory compliance (CDSCO + USFDA) essential for Indian manufacturers with international commercial ambitions; CDSCO approval is the domestic prerequisite |
[1,10,11,28] |
|
Market Access — USA |
USFDA Type II DMF [8] acceptance required for all APIs supplied in US-marketed drug products; PAI [42] mandatory for new API manufacturing sites; ongoing GDUFA III [41] compliance and annual DMF updates required |
US market access represents the highest commercial value regulatory investment for Indian API manufacturers; drives approximately USD 10+ billion in annual Indian pharmaceutical exports to the USA [1,2] |
[1,2,8,40,45] |
|
Export Revenue |
India pharmaceutical exports reached USD 25.4 billion (FY 2022–23) [1,2]; USA is the largest single export destination; API exports constitute approximately 40–37% of total export value; India supplies approximately 20–25% of global generic API volume |
Regulatory compliance is the fundamental prerequisite for export revenue sustainability; any regulatory action (Warning Letter, Import Alert) can immediately halt exports worth hundreds of millions of USD |
[1,2,48] |
|
Compliance Costs |
USFDA-related compliance costs (facility upgrades, validation, regulatory submissions, PAI preparation) estimated at USD 5–20 million annually for large API manufacturers; proportionally higher burden for MSMEs |
Government PLI scheme [69] for bulk drugs (INR 6,940 crore outlay); API parks development; MSME support programmes; industry consortium compliance platforms to distribute costs |
[69,48,73] |
|
Quality Improvement |
USFDA enforcement actions (Warning Letters, Import Alerts) against Indian facilities between 2014–2022 compelled systematic quality system overhauls [43,44]; revised Schedule M 2023 [11] raises domestic baseline for all manufacturers |
Long-term quality improvement driven by regulatory pressure benefits patient safety globally; manufacturers with ICH Q10 [20]-compliant QMS demonstrate improved batch success rates |
[7,12,14,43,44] |
|
Global Competitiveness |
India holds a dominant position in global generic API supply (approximately 20–25% by volume) [1,2]; USFDA approval status is a key competitive differentiator; manufacturers with strong dual CDSCO/USFDA regulatory compliance portfolios may command pricing advantages in regulated markets |
Compliance investment yields sustainable competitive advantage; though specific percentage premiums are difficult to substantiate without dedicated market research data [1,2] |
[1,2,48] |
|
Technology and Innovation Investment |
Compliance with ICH Q7 [18], Q8 [19], Q11 [21], data integrity, nitrosamine controls, and continuous manufacturing frameworks (ICH Q13 [23]) drives investment in advanced manufacturing technology, analytical instruments, and quality systems |
Quality-driven technology investment (LIMS, PAT, continuous monitoring) improves process efficiency and reduces batch failure rates, partially offsetting compliance costs |
[18,23,70] |
|
Government Support (India) |
PLI scheme for bulk drugs and KSMs (INR 6,940 crore over six years, 2020) [69]; dedicated API manufacturing parks; support for domestic manufacture of 40 critical APIs and key starting materials previously imported from China |
Government-industry alignment on reducing China dependence for critical API starting materials; strengthening domestic API manufacturing ecosystem through regulatory and financial support |
[69,73] |
|
Supply Chain Security |
COVID-19 pandemic (2020–2021) exposed API supply chain vulnerabilities; over-dependence on Chinese sources for key starting materials (KSMs) and intermediates for many strategic APIs |
Post-pandemic regulatory emphasis on supply chain diversification; FDA drug shortage prevention programmes; expedited approval pathways for alternative API sources; India's PLI scheme [69] directly addresses KSM supply vulnerability |
[69,73] |
The review shows that CDSCO and USFDA regulations which both use ICH quality guidelines demonstrate two distinct degrees of implementation because of their different levels of operational development and official processes and regulatory enforcement methods. The discussion brings together the most important results from the research which show their impact on Indian API producers and their governing bodies and the worldwide pharmaceutical distribution network.
The most consequential divergence identified is not in scientific philosophy—both systems are fundamentally aligned through ICH Q7 [18], Q8 [19], Q9 [17], Q10 [20], Q11 [21], and The ICH quality framework banks on its extensive scope yet needs to create specific procedural requirements which require formal documentation. The USFDA regulatory system delivers comprehensive public documentation to API manufacturers which explains all stages of their submission and review process together with the detailed requirements for different DMF types. [8,40], starting material designation criteria [9], eCTD specifications [29,20], nitrosamine control strategy [36], and fee structures [41]. CDSCO operates its procedures through fewer established rules which require manufacturers to make their own regulatory decisions without available guidance about DSSM designation and post-approval change classification and eCTD specifications. The practical compliance difficulties which this review discovered stem from the existing asymmetry. [15,46].
The results of the data integrity findings are particularly relevant. The continued trend of data integrity Warning Letters issued to Indian pharmaceutical manufacturers between 2014 and 2022, identified by peer-reviewed research [43, 44] as the most common type of compliance failure, indicates not a lack of scientific understanding but a lack of quality culture, computer system validation, and audit trail management. CDSCO's 2022 Data Integrity Guidelines [12], and the implementation of ALCOA principles in Schedule M 2023 regulations [11], represent an important regulatory response to this challenge. The apparent trend of a decline in enforcement actions related to data integrity after 2019 [43, 44] indicates that continued pressure from USFDA regulators and quality improvement initiatives by CDSCO regulators may be resulting in quality improvements—an observation consistent with the pharmaceutical regulatory literature [46,47].
The DSSM designation divergence described in Sections 4.4 and 5.4 is arguably the least appreciated but potentially significant operational problem for Indian API manufacturers seeking simultaneous registration from both CDSCO and USFDA. The implication of the finding that a single CTD Module 3.2.S dossier, intended for CDSCO compliance, is systematically non-compliant for FDA DSSM evaluation unless selection of the starting material was intended from the onset for FDA's DSSM requirements [9,21] is significant for API development strategy, investment in synthetic chemistry evaluation, and project timeline. The global regulatory science community would benefit from equivalent DSSM-specific guidance from CDSCO, which would also benefit manufacturers, regulators, and patients.
This comprehensive narrative review has offered an in-depth comparative analysis of the regulatory systems for API registration and Drug Master File submission for the Indian subcontinent (CDSCO) and the United States (USFDA), covering 22 key regulatory aspects for both the jurisdictions under consideration, including three unique aspects (DSSM designation standards, CEP recognition policy, and post-approval change comparison), which were not previously covered by the existing body of comparative reviews. This has established the fact that both the systems are distinct from each other, even though both share the same ICH quality guideline standards.
Some of the key similarities between CDSCO and USFDA are as follows: Their shared commitment to the ICH Quality Guidelines, i.e., Q7 (API GMP) [18], Q8 (Pharmaceutical Development) [19], Q9 (Quality Risk Management) [17], Q10 (Pharmaceutical Quality System) [20], and Q11 (API Development) [21], as a scientific and technical basis; the requirement for a comprehensive pharmaceutical dossier with a CTD Module 3.2.S; the requirement for stringent impurity profiling in compliance with ICH Q3A [25], Q3C [26], Q3D [27], and M7 [28], and the growing adoption of nitrosamine impurity control in response to the global valsartan impurity scandal [36, 37]. DMF systems are used as the main API registration vehicle in both countries, on-site GMP facility inspections are a prerequisite for approval in both countries, and risk-based approaches are increasingly used in the regulatory process in both countries for prioritization of inspections and reviews.
Essential differences stem from divergent paths in institutional development, resource allocation, and a differing approach in regulation. The USFDA has a highly formalized, digitally enabled, and transparent framework with mandatory use of eCTD (2017-2018) [29, 20], a highly codified five-type DMF categorization [8], OPQ quality review systems [39], highly stringent DSSM designation criteria (2022) [9], comprehensive PAI programs [42], ALCOA+ data integrity compliance [7], and review timelines with statutory GDUFA III performance commitments [41]. In contrast, CDSCO, with a highly dynamic SUGAM portal-based modernization process [13], Schedule M 2023 (G.S.R. 228(E) dated 28 March 2023) [11], PIC/S observer status [14], and CDSCO Data Integrity Guidelines 2022 [12], lacks highly codified DMF standards [34], a hybrid approach with digital and paper-based submission, a two-tiered federal-state structure, and, most critically, lacks DSSM-specific [9] or SUPAC-type guidelines, which creates a high degree of ambiguity for dual-registered manufacturers.
It is strategically crucial for the global pharmaceutical industry to comprehend and navigate both frameworks. The CDSCO-USFDA regulatory relationship directly affects the security of the global drug supply and patient access to reasonably priced medications because India is the world's largest supplier of generic APIs, accounting for 20–25% of global generic API volume [1,2]. API manufacturers that make proactive investments in regulatory compliance infrastructure, quality system excellence, and regulatory science expertise will be rewarded by future regulatory evolution, which will be fueled by mandatory global eCTD adoption [29,20], AI-assisted regulatory review [49], ICH Q13 continuous manufacturing frameworks [23], stricter nitrosamine and mutagenic impurity controls [28,36], and potential India-USA inspection information sharing after PIC/S full membership [14,31].
Manufacturers who create eCTD-ready submission infrastructure [29,20], ALCOA+-compliant data governance systems [7,12], and ICH Q11-aligned starting material selection strategies [21] will be in a position to effectively navigate both the CDSCO and USFDA frameworks, gaining access to the combined value of two of the most commercially significant pharmaceutical markets in the world.
The narrative review contains multiple limitations which readers must keep in mind when they evaluate its results. First, the literature selection process for this narrative review showed evidence of selection bias because the researchers selected sources which received high citation rates or which scholars widely regarded as accessible. Second, the rapidly evolving nature of pharmaceutical regulation means that specific guidance documents, fee schedules, and regulatory requirements cited may be updated after the March 2025 literature search cutoff; readers should verify current requirements directly with CDSCO (cdsco.gov.in) and USFDA (fda.gov) official sources. Third, timeline estimates in Tables 4 and 8, while grounded in CDSCO Annual Report 2022-23 [32] and GDUFA III performance data [41,45], The timeframes shown in this table represent typical ranges which do not provide 100% accuracy for actual durations; the time it takes to complete applications depends on three factors which are the quality of the dossier and the complexity of the API and the current workload of the regulatory agency. The review examines small-molecule chemical APIs while it excludes all biological APIs and biosimilar APIs and advanced therapy medicinal products. The regulatory comparisons presented in this document show simplified versions which maintain the necessary elements of several complex regulatory systems; practitioners should consult current primary regulatory guidance for specific compliance decisions.
REFERENCES:

Pranjali Lingalwar, Omkar Shivalkar, Amod Patil*, Pritam Jain, Nitin Haswani, CDSCO Versus USFDA Regulatory Systems for API Registration and DMF Submission: A Comparative Review of Harmonisation, GMP Compliance, And Regulatory Evolution, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 1981-2018. https://doi.org/10.5281/zenodo.21284489
 10.5281/zenodo.21284489
10.5281/zenodo.21284489