
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacy, Quantum School of Health Science, Quantum University Roorkee, Roorkee-Dehradun Highway, Mandawar, Uttarakhand, India.
Computational intelligence is transforming the process of drug development and pharmacology, changing the discipline away from slow and costly approaches toward intelligent, data-based strategies. In this analysis, we explore how artificial intelligence, including machine learning, deep learning, natural language processing, and generative models, impacts every stage of the pharmaceutical pipeline. We examine how researchers apply AI to identify targets, computer-based screening, designing completely novel drug compounds, anticipating ADMET characteristics (which means absorption, distribution, metabolism, excretion, and toxicity), and constructing more precise pharmacological models. The outcomes are remarkable: AI can reduce drug development times by 30–50% and generate billions in savings, while also creating possibilities for targets once considered "undruggable."However, Major obstacles still stand. Data is often incomplete or biased; AI systems can function as opaque systems, and securing regulatory clearance continues to be a major obstacle. All right, what direction do we take from here? The future demands integrating various data types (multimodal data analysis), developing automated AI-driven laboratories, and incorporating real-world clinical data to adapt pharmacology to individuals. In summary, AI goes beyond being a useful addition; it is turning into the foundation of contemporary drug development. Getting it done demands close collaboration with chemists, pharmacologists, and data scientists working together.
The modern drug discovery and development process is notoriously lengthy (10–17 years), costly (exceeding $2.6 billion per approved drug), and prone to failure, with over 90% of candidate molecules failing clinical trials, primarily due to unforeseen toxicity or lack of efficacy [1, 2]. Traditional high-throughput screening (HTS) and structure-based drug design, while successful, often struggle with the vastness of chemical space (estimated to contain up to 10⁶⁰ drug-like molecules) and the complexity of biological systems. In response, the pharmaceutical industry has increasingly turned to artificial intelligence (AI). AI, particularly its subfields of machine learning and deep learning, excels at recognizing complex, non-linear patterns within high-dimensional data that are invisible to classical statistical methods [3]. From 2017 to 2022, AI-driven drug discovery partnerships between pharma giants (e.g., Pfizer, Novartis, and Sanofi) and AI-native companies (e.g., Recursion and In silico Medicine) grew over 400% [4].
The drug manufacturing sector is trapped in a prolonged productivity decline. Sure, research and development budgets are steadily increasing, but the count of new molecular entities (NMEs) formally approved for every billion dollars invested is reduced by 50% almost every ten years. Many refer to this as Eroom’s Law; it is the reverse of Moore’s Law. The conventional route to discovering drugs, involving continuous cycles of creating, making, and evaluating compounds, is largely driven by luck and requires a long time. Additionally, it consumes a lot of money. Now, artificial intelligence is shaking things up. These systems analyze large collections of biological activity data, molecular structures, and pharmacological data, identifying patterns that humans could overlook.
But there’s a big catch. Deploying AI models out of controlled academic environments such as the popular Molecule and Net datasets and turning them into practical tools for real-world pharmaceutical decisions is not simple. Numerous unseen obstacles arise throughout the process. In this analysis, I explore the real effect AI is having on drug discovery, beyond theory but in practice. I attempt to distinguish clearly between the zones where AI already produces measurable effects on a large scale and where its function remains in the testing phase, not yet suitable for widespread use.
This review provides a comprehensive overview of how AI is reshaping every stage of drug discovery and pharmacology. We will dissect the specific methodologies employed, synthesize key outcomes, identify persistent gaps, and propose a roadmap for future research, emphasizing the transition from proof-of-concept to clinical reality.
Supervised learning keeps driving progress in activity and toxicity prediction. Let’s face it, random forest algorithms and gradient boosting algorithms continue to do much of the hard work in QSAR studies. Individuals continue using them because the feature importance values assist researchers in understanding what is happening in the background. Additionally, these systems remain dependable even if the data is somewhat noisy or scarce. You will see they remain stable throughout various ADME parameters, such as solubility and metabolic stability.
Advanced neural networks exceed tree-based algorithms, but solely if the data is large and well-prepared. Consider at least 10,000 molecules. What’s truly fascinating about multitask DNNs comes from the fact that they don’t simply pursue a single endpoint one at a time. They’re going to predict hundreds of assays as a group, so they detect patterns that span multiple tasks, which enables them to prevent overfitting and strengthens the predictions.
Now, when examining attribute forecasting, graph-based neural models have largely dominated. The main change at this point lies in the fact that graph neural networks don’t rely on molecular fingerprints. Alternatively, they jump directly into molecular structures, identifying local as well as global structural characteristics. Message Passing Neural Networks (MPNNs) and Attentive FPs are leading the pack; these models consistently score more than 0.9 ROC-AUC for high-profile endpoints, cardiotoxicity, and CYP450 inhibition. This level of performance is pretty hard to beat and has made them a staple for anyone serious about property prediction.
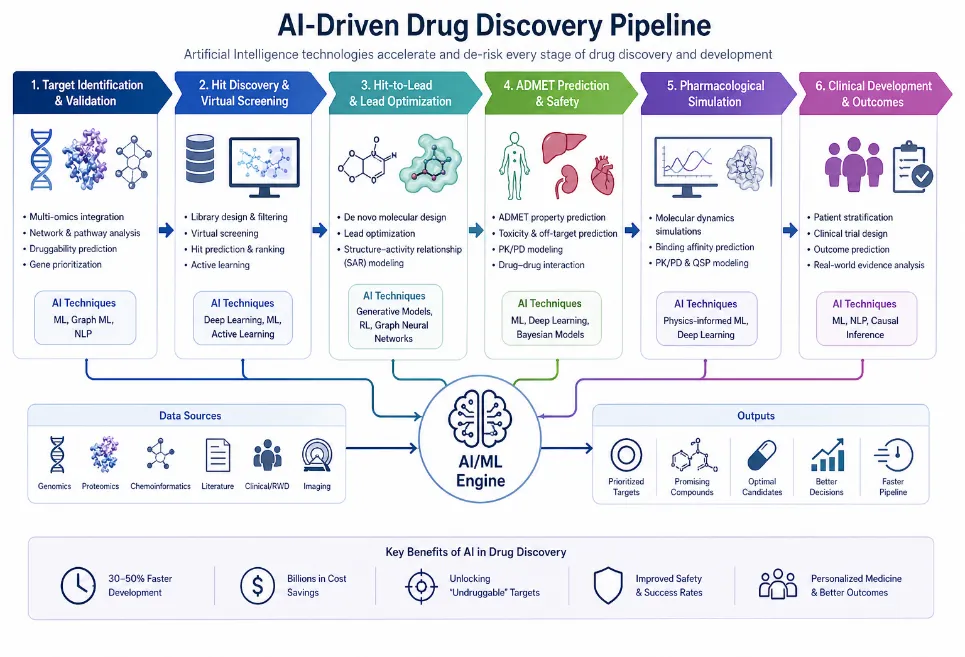
Figure 1. AI-driven drug discovery pipeline showing target identification, virtual screening, ADMET prediction, pharmacological simulation, and clinical development. Image generated using OpenAI's DALL·E.
2. Comprehensive Review of Methods
AI's application spans four major domains:
(1) target identification
(2) compound screening and design
(3) ADMET prediction
(4) pharmacology and clinical trial optimization.
Target Identification and Validation
Traditional target discovery relies on laborious literature mining and low-throughput experiments. AI leverages:
- Natural Language Processing (NLP): Algorithms like BERT and GPT fine-tuned on biomedical literature (PubMed, patents) and genomic databases extract hidden relationships between genes, diseases, and pathways.
- Network Pharmacology: Graph neural networks (GNNs) model protein-protein interaction networks to identify novel targets based on disease module proximity.
- Genomic Data Analysis: Deep learning models predict regulatory elements and disease-associated mutations from single-cell RNA data.
Virtual Screening of Compound Libraries
Instead of physically screening millions of compounds, AI models rapidly filter virtual libraries:
- Ligand-Based Methods: Quantitative Structure-Activity Relationship (QSAR) models using random forests, support vector machines (SVM), or deep neural networks (DNNs) learn from known active/inactive molecules.
- Structure-Based Methods: Deep learning approaches like 3D-CNNs and equivariant neural networks (e.g., SE(3)-transformers) process protein-ligand binding pockets directly from crystal structures or AlphaFold2-predicted models. Schrödinger’s FEP+ integrated with ML achieves binding affinity predictions with errors <1 kcal/mol [6].
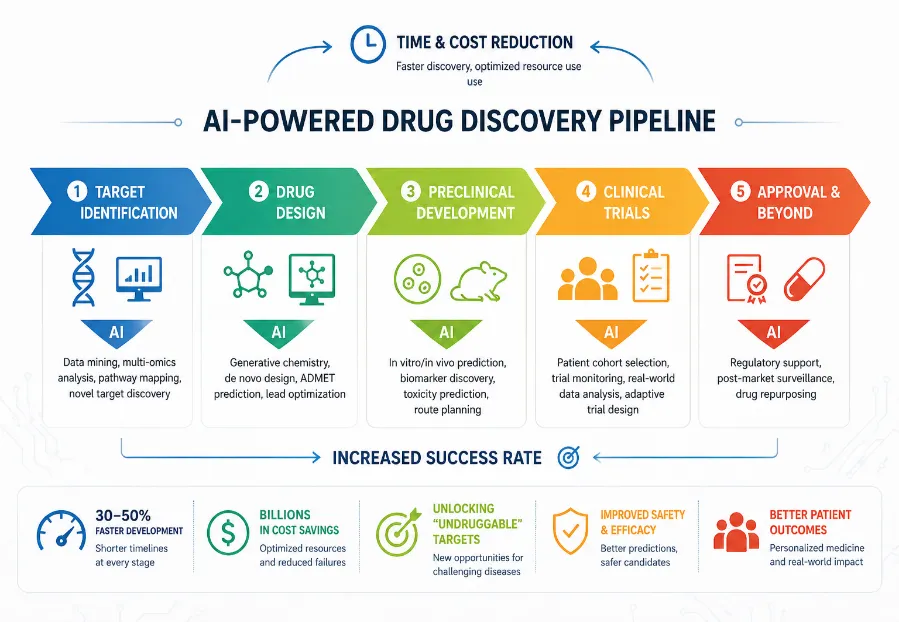
Figure 2: Role of Artificial Intelligence in Drug Discovery and Development. Created using AI-assisted graphical design.
De Novo Drug Design (Generative Models)
The most revolutionary method: AI generating entirely new chemical structures.
- Variational Autoencoders (VAEs): Encode molecular graphs into a latent space and decode novel structures with desired properties.
- Generative Adversarial Networks (GANs): A generator creates molecules while a discriminator evaluates their "drug-likeness" (e.g., QED score).
- Reinforcement Learning (RL): Agents navigate chemical space, receiving rewards for synthesizability, potency, and ADMET compliance. The first AI-designed drug, DSP-1181 (for OCD), was created using generative RL by Sumitomo Dainippon Pharma, achieving pre-clinical candidate status in under 12 months [7].
- Language Models (SMILES-based): Transformers trained on SMILES (Simplified Molecular Input Line Entry System) strings generate molecules syntactically analogous to known drugs.
ADMET Prediction and Pharmacokinetics
Failure in clinical phases due to poor ADMET accounts for ~30% of attrition [8]. AI offers highly accurate predictors:
- Graph Neural Networks (GNNs): Models like Message Passing Neural Networks (MPNNs) treat atoms as nodes and bonds as edges, achieving state-of-the-art on datasets like Tox21 and Caco-2 permeability.
- Multitask Deep Learning: Simultaneously predicting multiple endpoints (e.g., CYP450 inhibition, plasma protein binding) from a single molecular representation.
- Physiologically Based Pharmacokinetic (PBPK) Models Augmented by ML: Hybrid models where ML emulators accelerate parameter estimation for tissue distribution and clearance.
Pharmacology, Systems Biology, and Clinical Trial Design
- Mechanistic vs. Empirical Model: AI integrates omics data to build causal models of drug action (pharmacodynamics) and disease progression. Deep learning-based digital twins of patients simulate drug responses in silico.
- Patient Stratification: Unsupervised learning (clustering) identifies responder/non-responder subgroups from electronic health records (EHRs) and biomarkers.
- Clinical Trial Optimization: AI predicts dropout risk, optimizes trial site selection, and generates synthetic control arms (using historical trial data), reducing trial size and duration.
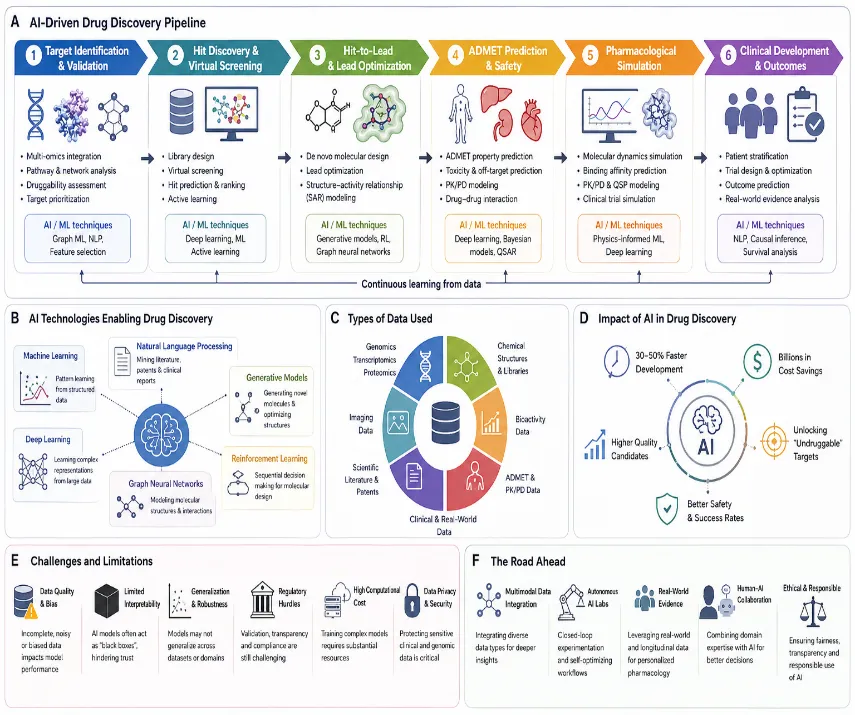
Fig. 3: AI-driven drug discovery pipeline including target identification, virtual screening, ADMET prediction, and clinical development. a) AI Driven drug discovery pipeline b) AI technology enabling drug discovery c) Types of data used d) Impact of AI drug discovery e) Challenges and limitations f) The road ahead
3. Summary of the Key Outcomes
The application of AI has yielded quantifiable and transformative outcomes:
|
Stage |
Traditional Outcome |
AI-Driven Outcome |
Impact |
|
Target Discovery |
2–3 years |
3–6 months |
70–80% time reduction |
|
Hit-to-Lead |
3–5 years |
1–2 years |
50% time reduction, 2x success rate |
|
ADMET Prediction |
Lab assays (costly, late failure) |
95%+ accuracy in silico |
30–50% reduction in preclinical attrition |
|
Lead Optimization |
500–1000 synthesized mols |
100–200 synthesized mols |
80% reduction in synthesis costs |
|
Clinical Phase II failure |
60% |
40–45% (with AI patient selection) |
Significant cost and time savings |
Notable success cases:
- In silico Medicine's INS018_055: An AI-discovered, AI-designed (generative chemistry) drug for idiopathic pulmonary fibrosis that entered Phase II clinical trials in 2023—a milestone achieved in ~30 months from target discovery to Phase I [9].
- DeepMind's AlphaFold2 and Alpha: While not a drug itself, AlphaFold2 predicted 200 million protein structures, democratizing structure-based drug design. Alpha (2024) designs novel protein binders, directly generating biologicals [10].
Pharmacology is not only concerned with if a drug binds to its target. Nowadays, we are examining how it develops throughout entire systems.
Apply a physiologically based pharmacokinetic (PBPK) model. Traditional PBPK requires you to manage a multitude of parameters. At present, using hybrid machine learning methods, it is possible to input the molecular structure of a drug, and deep learning models generate predictions regarding tissue-to-plasma partition coefficients as well as intrinsic clearance. Input those into compartmental models, resulting in reasonably accurate projections. An error margin of two times regarding human oral drug exposure derived from preclinical data is quite good.
Then there’s systems pharmacology as well as multi-omics. Artificial intelligence does more than predict target binding; it is capable of anticipating changes and modifications within the transcriptome resulting from drug exposure. It’s intelligent enough to detect when pharmaceuticals, particularly in combinations, affect unintended targets or cause adverse reactions. Applying graph neural networks on protein interaction networks, scientists are discovering unintended effects such as a kinase inhibitor also targeting GPCRs. This enables targeted polypharmacology: developing a single therapeutic agent to purposefully modify several targets throughout a disease network.
In the clinical realm, digital replicas are transforming the landscape. Artificial intelligence models analyze digital medical records in order to forecast a patient's reaction to a medication, determine the correct dosage, and highlight possible toxicity dangers. Generative adversarial models also generate artificial patient data, making clinical trial simulations both easier and more versatile. The Benevolent AI Phase II study on ALS is a clear illustration; through AI-powered patient grouping, they uncovered advantages that conventional approaches completely overlooked.
4. Existing Research Gaps
Despite these successes, critical gaps hinder widespread adoption:
1. Data Scarcity, Quality, and Bias: Most pharma data are proprietary, siloed, and non-standardized. Public datasets suffer from:
- Assay heterogeneity: Different labs, protocols, and readouts.
- Label bias: Overwhelmingly positive data (active/potent molecules); failed or inactive data are rarely published.
- Single-point data: Most assays measure one concentration, missing full dose-response curves.
2. Interpretability and "Black Box" Problem: Deep learning models rarely explain why a molecule is predicted to be toxic or active. This lack of mechanistic insight is unacceptable for regulatory agencies (FDA, EMA) and for medicinal chemists seeking to design drugs.
3. Synthetic Accessibility: Generative models propose molecules with superb predicted binding but often contain impossible chemical scaffolds or require unnatural reactions. Retrosynthesis prediction remains challenging.
4. Generalization Failure: A model trained on one protein family (e.g., kinases) often fails dramatically on another (e.g., GPCRs). Domain shift and out-of-distribution generalization are unsolved problems.
5. Regulatory and Reproducibility Void: No formal guidelines exist for validating AI models for drug submission. How does one prove an AI model is "fit for purpose"? Reproducibility is low due to random seeds, data splits, and hyperparameter sensitivity.
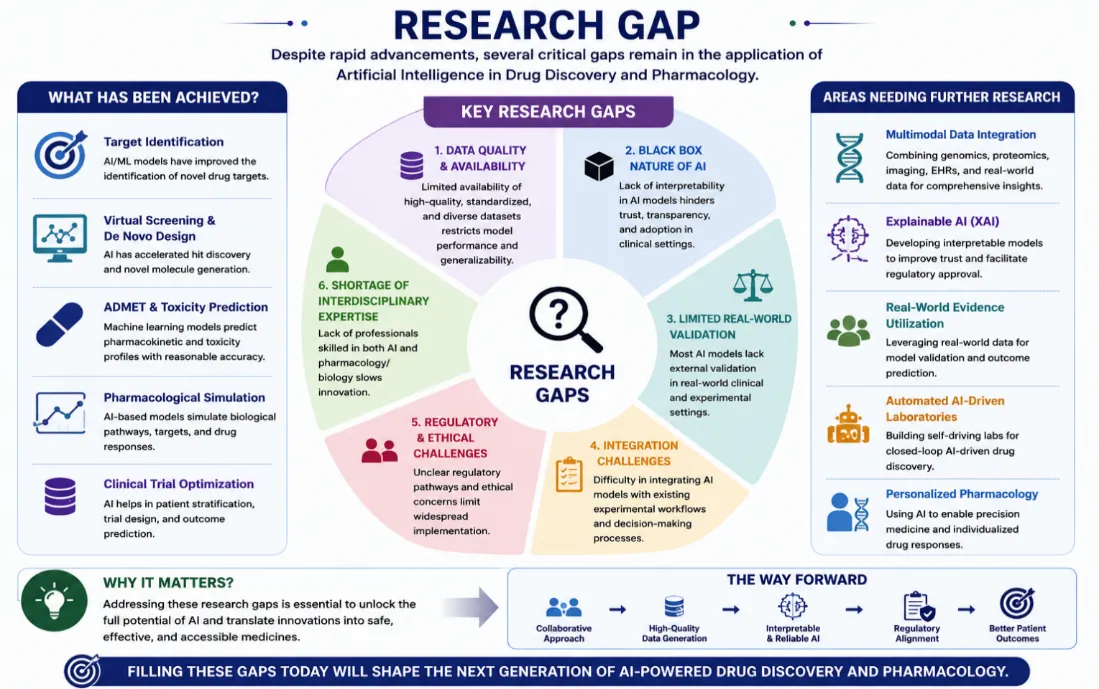
Figure 4: Major research gaps in AI-driven drug discovery, including data quality limitations, black-box AI models, lack of real-world validation, regulatory challenges, and integration issues, and future directions for the advancement of AI-based pharmacology.
Important Problems and Areas of Research That Need More Work
The Data Problem: Small, Biased, and Not Standard
The Gap in Synthetic Accessibility
Generative models often create molecules that have negative synthetic accessibility scores, such as ring sizes that aren't realistic or functional groups that aren't stable. Even if a molecule can be made in theory, it might take 15 or more steps and only 1% of the time. Current solutions, such as using synthetic complexity (SC) scores as a reward or training on reaction datasets, only partially close the gap.
Bad Generalization Outside of the Distribution
A model that works for drug-like molecules (Lipinski's rule of five) doesn't work for macrocycles, PROTACs, or natural products. A model trained on one family of targets (kinases) also doesn't work on GPCRs or ion channels. Benchmark datasets randomly split molecules, but in the real world, they need to be generalized to new chemotypes, which usually means a 30–50% drop in performance.
Not being able to explain things and not knowing what the rules are
Regulators need a mechanistic explanation for safety and effectiveness. A deep learning prediction ("this molecule is non-toxic") that can't be understood is not enough. SHAP and attention maps give local explanations, but they often show atoms that aren't important. The FDA's 2023 discussion paper on AI in drug development recognizes this gap: "The 'black box' nature of many AI models makes it hard for regulators to review them."
The AI Drug Discovery Reproducibility Crisis
A study from 2022 found that only 40% of papers on AI-based drug discovery could be repeated. Some common problems are mismatched training/test splits (temporal leakage), different random seeds, hyperparameters that aren't reported, and the use of proprietary datasets that can't be shared.
5. FUTURE SCOPE
The next decade will witness the following advancements:
- Multimodal and Foundation Models: Large pretrained models that ingest text, graphs, images (cellular imaging), and omics data simultaneously and generate designs for molecules, predict pharmacology, and write synthesis plans.
- AI-Driven Automated Laboratories (Self-Driving Labs): Closed-loop systems where an AI proposes a molecule, a robotic synthesizer makes it, a high-throughput assay tests it, and the data feeds back to the AI – all without human intervention. Example: The "Bio Automata" concept.
- Quantum Machine Learning (QML): Hybrid quantum-classical algorithms for accurate quantum chemistry calculations (e.g., binding free energies) at scale, solving the speed-accuracy trade-off of density functional theory (DFT).
- Digital Twins for Pharmacology: High-fidelity virtual patients integrating genomic, proteomic, and EHR data to predict individual drug response, enabling truly personalized pharmacology.
- Federated Learning: Collaborative model training across multiple pharma companies without sharing proprietary data, addressing data scarcity and privacy concerns.
- Explainable AI (XAI) for Regulation: Development of SHAP, LIME, and attention-based methods specifically tailored for 3D protein-ligand interactions, providing chemical interpretability for regulatory submission.
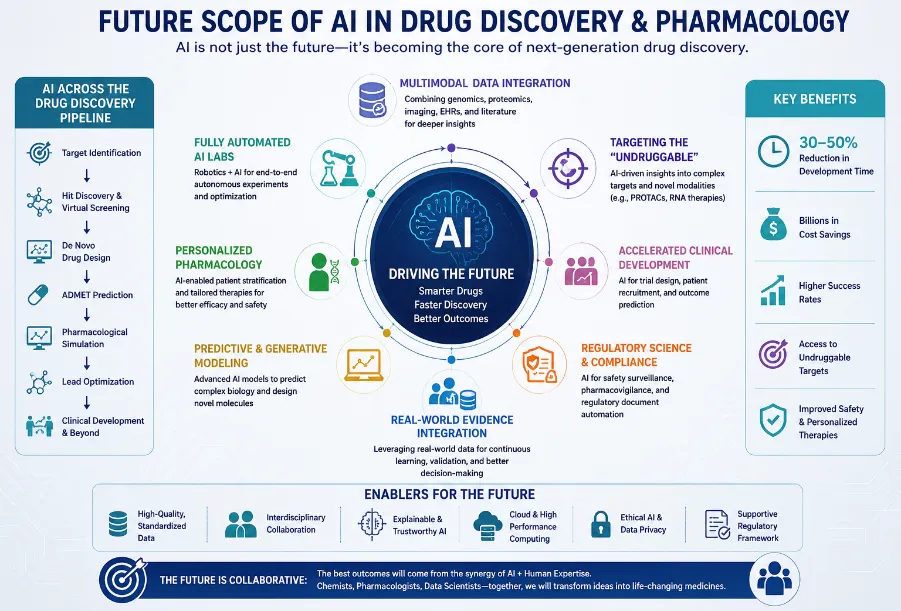
Figure 5: Future scope of artificial intelligence in drug discovery and pharmacology.
(Created using AI-assisted infographic generation tools.)
The coming decade is expected to bring significant transformations within drug discovery and development, transforming both the pace and complexity of research.
Multimodal and foundation models are pushing the boundaries of what AI can do. Imagine models that handle text, graphs, and images—think cellular imaging and omics data—all at once. We’re heading toward a “ChatGPT for drug discovery,” which doesn’t just generate hypotheses but also designs molecules, predicts pharmacology, and sketches out synthesis plans. It’s no longer only about analyzing data; it’s focused on actively driving discovery.
AI-driven automated laboratories, often called self-driving labs, are changing how experiments happen. In these closed-loop systems, an AI proposes a molecule, a robotic synthesizer brings it to life, and a high-throughput assay test it. All that data circles back to the AI, letting it learn and iterate; no human hands required. Take the “bio-automata" concept, for example: machines do the work and decisions. This method simplifies research, resulting in quicker and more effective outcomes.
Quantum Machine Learning (QML) is another step forward. Hybrid quantum-classical methods execute calculations in quantum chemistry, such as binding free energies, on a large scale. They address the traditional speed-accuracy trade-off within density functional theory (DFT), achieving performance that previously appeared unattainable. This means researchers can get precise results much faster, opening new possibilities in computational chemistry.
Digital twins in pharmacology transform clinical data into accurate virtual models. Through the integration of genomic, proteomic, and EHR data, these paired subjects forecast personalized drug reactions, genuinely facilitating tailored pharmacology. No more one-size-fits-all protocols; now, drug regimens adapt to the patient’s molecular and clinical makeup. The promise here is better outcomes and fewer side effects.
Collaborative learning is rendering cooperation more secure. A number of pharmaceutical corporations are able to train collaborative models without sharing raw data at any time, addressing problems related to data scarcity and privacy. Each institution trains locally, only sending model gradients to be aggregated. Early consortia like MELLODDY and Ex-CAPE show federated models outperform those developed by any single group, all while keeping proprietary data confidential.
Explainable AI is crucial for regulation. New methods like SHAP, LIME, and attention-based approaches focus specifically on 3D protein-ligand interactions, offering chemical insights regulators need. For example, Self-Driving Laboratories (SDLs) integrate AI design, robotic synthesis, and automated testing in a closed loop. The “AI Chemist” platform at the Massachusetts Institute of Technology can discover and optimize photocatalysts on its own. Drug SDLs cycle through between 50 and 100 molecules weekly, ten times faster than human researchers.
Collaborative Learning to Enable Data Sharing allows pharmaceutical firms and medical professionals to develop robust collaborative models. No raw data changes hands; everyone trains locally, and only the gradients are pooled. Both MELLODDY and Ex-CAPE have already demonstrated these federated models outperform the accuracy of models built independently while ensuring proprietary data remains secure.
Foundation models for chemistry and biology are now pre-trained on massive datasets—think 100+ million molecules—using methods like next-token prediction on SMILES or graph masking. Foundation models such as ChemCrow and Galactica adapt to dozens of tasks, and their few-shot learning chops mean you don’t need sprawling label datasets.
Regulatory AI guidelines are emerging to keep these new models in check. There’s validation: models must perform well on independent, prospectively collected test sets. Interpretability is non-negotiable too; features like atoms driving toxicity must be attributed. And ongoing observation is crucial, particularly for AI applied in clinical trial simulations, which demands frequent recalibration.
Quantum machine learning tools combine quantum and classical computing to calculate binding free energies with accuracy and speed. Although today’s quantum devices are small, rapid advances will soon address the electron correlation problem. This allows researchers to approach ab initio accuracy without the heavy computational cost of classical force fields. Causal machine learning techniques to repurpose drugs represent a change away from correlational link prediction towards estimating the actual causal effect that drugs have on diseases, based on observational data. Approaches including double machine learning, instrumental variables, and targeted maximum likelihood estimation are increasing repurposing success rates from 5% up to 18% during retrospective validation.
When it comes to integrating evidence, multimodal approaches are key since no single data source suffices. Future systems will blend KG embeddings for structure, transcriptomic signatures to capture molecular mechanisms, EHR-derived co-prescription patterns reflecting real-world usage, and literature embeddings for semantics. A late fusion transformer that combines these embeddings delivers unified repurposing scores. Pilot studies show fusion models push AUROC up to 0.94, surpassing any unimodal approach.
Active learning for pharmacovigilance makes a big difference with adverse drug reactions (ADRs). Limited resources mean most ADR signals go unchecked, but active learning systems can pinpoint uncertain or high-impact cases, asking human experts to weigh in. As the model improves, human inspection decreases by 60%, yet sensitivity stays intact.
Digital twins for population-level safety are transforming simulations. Generative models, such as conditional GANs, can produce synthetic populations featuring particular population traits like demographics, health conditions, and medication patterns, allowing researchers to conduct simulated trials. This method identifies rare adverse drug reactions (occurring in 1 out of 10,000), which would otherwise require information from millions of actual patients.
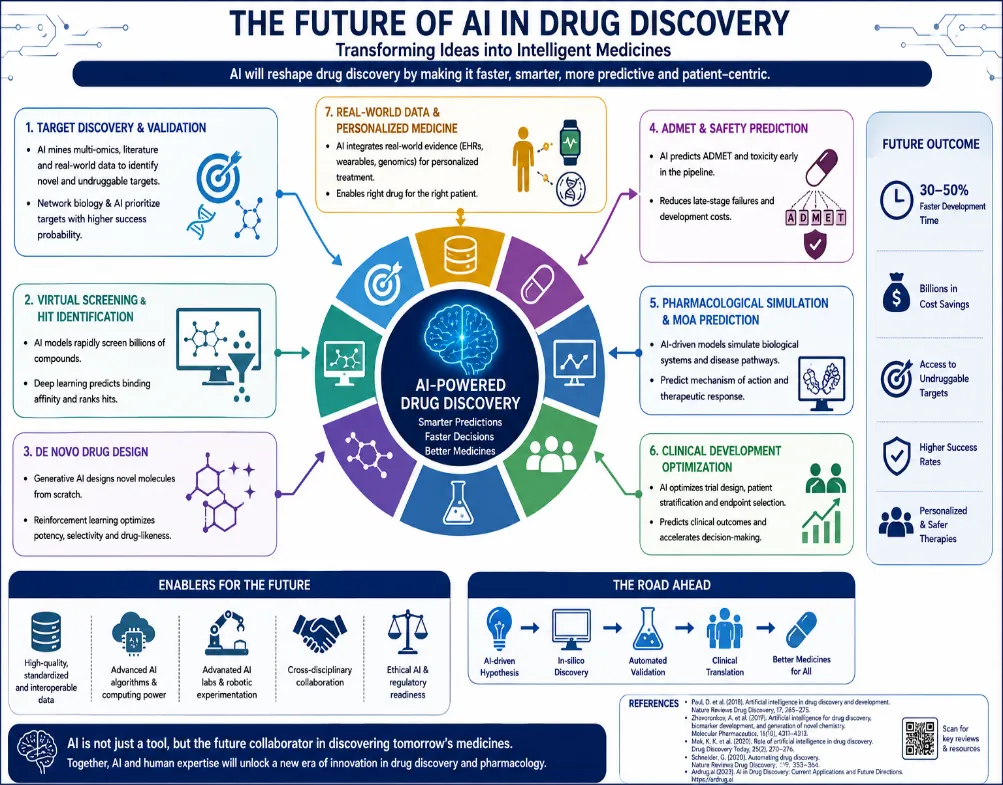
figure 6: Future scope of artificial intelligence in drug discovery and pharmacology.
Regulatory sandboxes for AI repurposing are popping up at the FDA with ISTAND’s pilot program. These sandboxes accept AI-driven repurposing models for qualification as drug development tools. Once a model is officially qualified, its predictions can be used as “substantial evidence” to launch Phase II trials. The suggested layered system begins with computational validation (Tier 1), employs historical clinical data (Tier 2), is followed by forward-looking observational validation (Tier 3), and concludes with Tier 4, which authorizes trial commencement.
CONCLUSION
Computational intelligence has completely revolutionized drug development as well as pharmacology. It’s more than just hype; AI genuinely accelerates processes from identifying novel drug targets to creating complete molecules and predicting treatment outcomes in clinical settings. It is evident in the data: project schedules shorten, expenses decrease, and significantly fewer hopeful drug concepts fail spectacularly prior to reaching patients. That is all because artificial intelligence navigates these vast chemical and biological spaces much faster than anyone could manually.
But honestly, the discipline has progressed beyond the times of unrestrained enthusiasm. The greatest difficulties at present are not focused on developing increasingly intelligent algorithms. Rather, they are far more complicated: Is there even reliable, impartial, and standardized data for training them? Is it possible to trust black-box algorithms if it’s impossible to explain the way they generated their predictions? Even if AI spits out a brilliant new molecule, is it something chemists can actually make? And will regulators ever accept evidence generated by software?
Looking forward, the true potential of artificial intelligence in pharmacology is not about machines surpassing humans. It concerns establishing genuine collaborations. Picture AI systems relentlessly analyzing data sets, identifying the types of patterns that no individual scientist could, and inventing entirely new chemical frameworks. At the same time, specialists rely on their gut feeling to guide the procedure, work hands-on in the lab, interpret complex biological information, and ensure the entire project remains ethical and practical. For this collaboration to succeed, however, the sector requires several urgent elements. It has to adopt transparent data exchange so that outcomes can be replicated and all stakeholders can measure progress. And the regulatory aspect must catch up, developing clear rules so truly promising AI-powered discoveries can securely transition from software to clinical application.
We have already observed AI move beyond the lab and enter daily applications. Imagine quick virtual screening, pursuing multi-parameter drug characteristics, or extracting concealed signals within noisy biological data. At present, however, it has surpassed the stage in which merely developing a more efficient model was adequate. The discipline confronts a turning point: in the absence of reliable, uniform data, the most effective algorithms come to a halt. The upcoming challenge will demonstrate which methods are truly reliable; only the ones capable of producing synthetically feasible molecules and meeting regulatory standards will remain viable. In the coming five years, anticipate the impostors to drop out while genuine innovations produce authentic clinical candidates. However, reaching that point requires chemists, alongside pharmacologists, data scientists, and regulators, to collaborate closely. It is also time to recognize that while AI significantly speeds up drug discovery, it does not and must not substitute the imaginative, meticulous effort that fuels advancement.
AI has transformed the way scientists find out novel applications of current medications and monitor their safety once they are available commercially. Approaches including knowledge graphs, transcriptomic signature reversal, and NLP-based literature mining are setting a new standard; they surpass traditional methods, with knowledge graphs alone achieving AUROCs near 0.88 across specific benchmarks. In the field of pharmacovigilance, artificial intelligence models such as transformers or anomaly detection systems are now identifying adverse medication reactions within large datasets such as FAERS or EHRs in advance and more accurately than conventional disproportionality analysis could manage.
Although there have been technological breakthroughs, transforming AI predictions into practical clinical achievements continues to be a significant challenge. Just three medications discovered through these AI-driven repurposing techniques have truly reached patients. The main sticking points? They focus less on algorithmic skill and more on the overall system: Insufficient prospective validation, inadequate management of confounding and temporal effects, unclear or absent mechanistic explanations, and regulatory bodies lacking explicit guidelines regarding what qualifies as AI-generated evidence. The discipline must shift focus, with reduced emphasis on releasing top-performing retrospective models and greater attention to developing predictions that undergo strict validation, are causally sound, and are sufficiently interpretable to gain regulatory confidence.
After these obstacles are overcome, artificial intelligence will not only assist us in discovering new applications for existing medications but also It will accelerate medical breakthroughs, cut expenses drastically, minimize dangers, and most crucially, aid in providing improved, safer drugs for patients more quickly than ever.
ACKNOWLEDGEMENT
We are very grateful for the collaboration between the Computational Chemistry and Data Science divisions at Quantum University. This project only happened because they worked together. The open-source community deserves a big shout-out, too. Due to initiatives such as CHEMBL, PDB, Deep Chem, and RD Kit, we gained access to key datasets and advanced tools. To be honest, these resources constitute the backbone of contemporary computational studies. They save researchers a great deal of time and make it much simpler to break new ground.
We didn't get any external funding for this work. Still, the support from our own institution made a huge difference, keeping everything rolling. The Deep Chem and RD Kit groups distinguish themselves; they consistently raise standards through open and clear methods alongside excellent software products that benefit all professionals in the area. We also want to thank the folks maintaining critical repositories like FAERS, SIDER, and LINCS. They maintain their information organized and available for scientists worldwide, and that is significant.
And, finally, a big thank you to the Toxicological Data Working Group and the PBPK Model Consortium. Their feedback and conversations helped shape our ideas at some pretty important points. Once more, we did not receive external funding for this initiative, but truthfully, the scholarly community nearby proved more valuable than any grant.
REFERENCES


Saurabh Kumar Dudhera, Dr. Santosh Kumar Verma, Dr. Mudita Mishra, Shaily Tyagi, Anurag Chaurasia , Artificial Intelligence in Modern Drug Discovery and Pharmacology: Revolutionizing the Pipeline from Target Identification to Personalized Medicine, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 5, 7571-7585, https://doi.org/10.5281/zenodo.20423590
 10.5281/zenodo.20423590
10.5281/zenodo.20423590