
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1 Applied Chemistry (Pharmaceuticals), Guru Nanak Dev University, Amritsar
2 Synthetic Chemistry Pharmaceuticals and Fine Chemicals, Department of Chemistry, University College Dublin, Ireland
3 Pharmaceutical Chemistry, Maulana Abul Kalam Azad University of Technology, Bharat Technology, West Bengal
Click chemistry, introduced by Sharpless and colleagues in 2001, has fundamentally transformed the way chemists and biologists construct molecules, probe biological systems, and develop therapeutics. Defined by its hallmark attributes—high yields, exquisite selectivity, mild reaction conditions, and compatibility with aqueous environments—click chemistry has evolved from an elegant synthetic concept into an indispensable technological platform. This comprehensive review systematically examines the mechanistic underpinnings and synthetic scope of the principal click reactions: copper(I)? catalyzed azide? alkyne cycloaddition (CuAAC), strain? promoted azide? alkyne cycloaddition (SPAAC), inverse? electron? demand Diels? Alder (IEDDA) reactions involving tetrazines and strained alkenes, Staudinger ligation, oxime and hydrazone conjugation, and thiol? ene additions. We critically evaluate their applications across the drug discovery pipeline—from fragment? based screening and combinatorial library synthesis to activity? based protein profiling (ABPP) and covalent drug design—and across the increasingly strategic arena of bioconjugation, encompassing antibody? drug conjugates (ADCs), site? specific protein modification, PEGylation, and radiolabelled imaging agents. Special attention is devoted to bioorthogonal click chemistry, which has enabled real? time molecular imaging in living organisms and tissue? selective prodrug activation. Emerging frontiers including sulfur(VI) fluoride exchange (SuFEx), photoclick chemistry, and click? to? release strategies are thoroughly discussed. We also address persistent challenges—copper cytotoxicity, pharmacokinetic liabilities of triazole? containing drugs, and linker stability in ADCs—and outline translational pathways toward clinical implementation. Collectively, this review underscores click chemistry's transformative role in bridging synthetic chemistry and molecular medicine, and charts a future where precision bioconjugation enables next? generation targeted therapies.
The synthesis of complex bioactive molecules has historically been constrained by multi step reaction sequences, low yields, harsh conditions, and poor compatibility with biological environments. The articulation of click chemistry by Sharpless, Finn, and Kolb in 2001 offered a paradigm shifting answer to these limitations [1]. The term 'click chemistry' described a set of reactions that are modular, wide in scope, give very high yields, generate only inoffensive by products, are stereospecific, simple to perform, and require no or minimal purification [1]. The founding philosophy was not to replicate nature's combinatorial ingenuity but to exploit a small number of near perfect reactions to generate vast chemical diversity from simple precursors.
Among the originally catalogued click reactions thiol ene additions, Diels Alder cycloadditions, nucleophilic ring openings of strained epoxides and aziridines, and multicomponent reactions—the copper(I) catalyzed azide alkyne cycloaddition (CuAAC), independently discovered by Sharpless and Meldal in 2002 [2,3], rapidly emerged as the quintessential click reaction. Its extraordinary reliability, chemoselectivity, and broad substrate tolerance made it uniquely positioned for integration into biological systems, material sciences, and pharmaceutical sciences.
The biological frontier of click chemistry was pioneered by Bertozzi and colleagues, who demonstrated that specific chemical reactions could be executed inside living organisms without perturbing the native biochemistry—a concept termed 'bioorthogonality' [4,5]. This breakthrough, together with Meldal's contribution to triazole forming reactions, was recognized by the 2022 Nobel Prize in Chemistry, awarded jointly to Sharpless, Bertozzi, and Meldal [74]. Bioorthogonal click reactions have since enabled the real time visualisation of glycans on cell surfaces [22], metabolic labelling of newly synthesised proteins [63], and intracellular drug activation in animal models [48].
In drug discovery, click chemistry has permeated virtually every stage of the pipeline. In target identification and validation, it underpins activity based protein profiling (ABPP), enabling the global mapping of enzyme active sites in complex proteomes [44,45]. In hit identification, CuAAC based fragment assembly accelerates the construction of enormous chemical libraries for high throughput screening (HTS) [57]. In lead optimisation, the triazole moiety confers favourable pharmacokinetic properties, resistance to metabolic degradation, and the capacity for hydrogen bond donation [16]. In targeted therapy, click bioconjugation provides the molecular glue for antibody drug conjugates (ADCs), PEGylated biologics, and site specifically radiolabelled imaging agents [39,41,50].
This review is organised to guide the reader through the mechanistic foundations of major click reactions (Section 2), their specific applications in drug discovery (Section 3), their role in bioconjugation strategies (Section 4), their deployment in imaging and diagnostics (Section 5), their integration into materials and drug delivery (Section 6), and the emerging frontiers of SuFEx, photoclick, and click to release chemistry (Section 7). We conclude by examining translational challenges, regulatory considerations, and prospective directions for the field.
MECHANISTIC FOUNDATIONS OF PRINCIPAL CLICK REACTIONS
Copper(I) Catalyzed Azide Alkyne Cycloaddition (CuAAC)
The CuAAC reaction couples an organic azide with a terminal alkyne to afford 1,4 disubstituted 1,2,3 triazoles with absolute regioselectivity. Prior to the discovery of copper catalysis, the uncatalyzed Huisgen [3+2] dipolar cycloaddition between azides and alkynes required temperatures exceeding 100°C and yielded mixtures of 1,4 and 1,5 regioisomers [13]. The introduction of Cu(I) not only provided regiocontrol but also reduced activation energy by approximately 11 kcal/mol, enabling quantitative conversion at room temperature in water miscible solvents [18,19].
Mechanistic investigations, corroborated by DFT computations and isotopic labelling studies, established that the catalytic cycle proceeds through a dinuclear copper intermediate [66]. The Cu(I) species, typically generated in situ by reduction of Cu(II) sulfate with sodium ascorbate, coordinates to the terminal alkyne, lowering its pKa and enabling metallacycle formation. This sigma,pi bis(copper) acetylide undergoes selective ring closure with the azide, proceeding through a six membered copper containing metallacycle before protodemetalation releases the 1,4 triazole product. Ligands such as THPTA, BTTES, and BTTAA accelerate the reaction further and significantly reduce copper mediated oxidative damage to DNA and proteins, extending CuAAC's utility to living cell experiments [21].
From a medicinal chemistry perspective, the 1,2,3 triazole ring is not merely a passive linker. Bioisosteric relationships with amide bonds and cis double bonds make triazoles attractive pharmacophores. The triazole's dipole moment (approximately 5 D), its hydrogen bond acceptor nitrogen atoms, and its metabolic stability confer favourable drug like properties [14,16]. Several clinically used drugs including rufinamide (antiepileptic) and tazobactam (β lactamase inhibitor) contain triazole moieties, validating the scaffold's medicinal relevance.
Strain Promoted Azide Alkyne Cycloaddition (SPAAC)
The cytotoxicity of copper at concentrations required for efficient CuAAC in cellular contexts motivated the development of copper free alternatives. SPAAC, pioneered by Bertozzi's group in 2004, exploits ring strain in cycloalkynes to achieve sufficient reactivity without metal catalysis [4]. Cyclooctyne, with approximately 18 kcal/mol of ring strain, reacts with azides at rates (k₂ ≈ 10⁻³ M⁻¹s⁻¹) that are orders of magnitude below CuAAC but sufficient for biological labelling applications given the low background of azides and strained alkynes in biological matrices [9].
Subsequent generations of strained alkynes dramatically improved reaction kinetics. Difluorinated cyclooctyne (DIFO) [5], dibenzocyclooctyne (DIBO) [23], bicyclononyne (BCN) [23], and biarylazacyclooctynone (BARAC) achieved second order rate constants of 0.1–1.0 M⁻¹s⁻¹, enabling labelling of sparse surface glycans in minutes. DBCO (dibenzocyclooctyne), commercially the most prevalent reagent, offers rates of approximately 0.3–1.0 M⁻¹s⁻¹ and has become the workhorse for in vivo bioorthogonal imaging, ADC synthesis, and nanoparticle functionalisation [24,54]. The SPAAC reaction is thermodynamically irreversible, highly selective, and tolerates physiological aqueous environments, making it the preferred click chemistry for in vivo applications.
Inverse Electron Demand Diels Alder (IEDDA) Ligation
The tetrazine ligation, independently reported by Fox and Weissleder groups in 2008 [6,7], represents the fastest bioorthogonal reaction known. Electron deficient tetrazines react with strained dienophiles—principally trans cyclooctene (TCO), norbornene, and cyclopropene derivatives—via inverse electron demand Diels Alder cycloaddition followed by retro [4+2] elimination of dinitrogen to yield stable dihydropyridazine or pyridazine products. The rate constants span an extraordinary range: norbornene reacts at ~1 M⁻¹s⁻¹, whereas optimised TCO tetrazine pairs achieve k₂ values of 10⁴–10⁶ M⁻¹s⁻¹, enabling sub second bioconjugation at nanomolar concentrations [25,26].
The unparalleled speed of tetrazine ligation is its defining advantage. At picomolar to nanomolar probe concentrations—relevant to PET imaging, where long biological half lives are undesirable—the IEDDA reaction remains efficient, enabling pretargeting strategies where an antibody TCO conjugate accumulates at the tumour before a small molecule tetrazine radiometal chelate is administered [51]. Design of tetrazines with tunable stability, hydrophilicity, and reactivity has been the subject of intensive synthetic investigation [60]. Monosubstituted tetrazines bearing pyrimidyl or pyridazyl groups achieve an optimal balance between stability in aqueous media and high reactivity, whereas disubstituted tetrazines with pyridyl substituents are more stable but slower.
Staudinger Ligation
The modified Staudinger ligation, introduced by Saxon and Bertozzi in 2000 [29], predates CuAAC based bioorthogonal chemistry and represents a historically significant milestone. In this reaction, an organic azide reacts with a triarylphosphine bearing an ester linked electrophilic trap; the initially formed aza ylide undergoes intramolecular cyclisation and hydrolysis to afford a stable amide linked product with release of phosphine oxide. The reaction is completely bioorthogonal, proceeds in aqueous environments, and was the first reaction used to image azide tagged glycans on living cells and in living mice [30].
However, the Staudinger ligation suffers from slow kinetics (k₂ ≈ 0.002 M⁻¹s⁻¹), susceptibility of the phosphine to air oxidation, and relatively large probe sizes, all of which limit its in vivo utility. Nonetheless, its absolute selectivity for azides makes it valuable in proteomics workflows where temporal resolution is less critical, and traceless variants that do not append the phosphine oxide to the product have been developed for specific applications [31]. For live animal studies, it has largely been superseded by SPAAC and IEDDA.
Oxime and Hydrazone Ligations
Oxime and hydrazone bond formation—condensation of an aldehyde or ketone with a hydroxylamine or hydrazide, respectively—predates click chemistry as a bioconjugation strategy but has been formally incorporated into the click framework for its reliability and chemoselectivity [32,33]. These reactions are particularly useful in protein and antibody labelling, where ketone or aldehyde handles can be introduced by genetic encoding of unnatural amino acids or by enzymatic oxidation of engineered glycosylation sites. Rate constants are modest (k₂ ~ 10⁻⁴–10⁻² M⁻¹s⁻¹ at physiological pH) but can be dramatically enhanced by nucleophilic aniline catalysis [32]. Hydrazone bonds are reversible under acidic conditions, a property exploited in pH responsive linker designs for ADCs where selective release in the acidic endolysosomal compartment of cancer cells is desired.
Thiol Ene and Thiol Michael Additions
Thiol ene click reactions—the addition of thiols across carbon carbon double bonds—proceed via radical (UV initiated) or ionic (base/nucleophile catalyzed, thiol Michael) mechanisms [35,68].
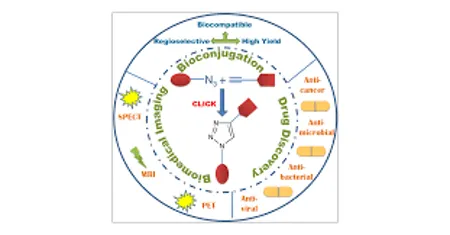
FIG.1 CLICK CHEMISTRY
Both pathways share the essential click attributes: quantitative yields, regioselectivity, and aqueous compatibility. Radical thiol ene reactions are particularly valued in materials science for spatiotemporally controlled hydrogel crosslinking and surface patterning under UV irradiation, while thiol Michael additions—involving thiols and electron poor alkenes such as maleimides, acrylates, and vinyl sulfones—are the cornerstone of site specific ADC conjugation via inter chain disulfide reduction and maleimide quenching [35,68].
Conditions, and Applications
|
Reaction Type |
Reagents |
Conditions |
Yield (%) |
Key Applications |
|
CuAAC |
Azide + Terminal Alkyne |
Cu(I) catalyst, RT, H₂O/EtOH |
85–99 |
Drug conjugation, PET tracers, polymer synthesis |
|
SPAAC |
Azide + Cyclooctyne |
Cu free, physiological |
70–95 |
Live cell imaging, in vivo bioorthogonal labelling |
|
Inverse demand DA |
Tetrazine + TCO/norbornene |
No catalyst, fast kinetics |
80–99 |
Pretargeting radioimmunotherapy, protein labelling |
|
Thiol–ene |
Thiol + Alkene |
UV or radical initiator |
75–95 |
Hydrogel fabrication, surface functionalisation |
|
Staudinger Ligation |
Azide + Phosphine |
Aqueous, no metal |
60–85 |
Proteomics, glycan imaging |
|
Oxime/ Hydrazone |
Aldehyde/Ketone + Oxyamine/Hydrazide |
Acidic pH, aniline catalyst |
70–92 |
Antibody–drug conjugates, peptide cyclisation |
CuAAC = copper(I) catalyzed azide alkyne cycloaddition; SPAAC = strain promoted azide alkyne cycloaddition; DA = Diels Alder; RT = room temperature; TCO = trans cyclooctene.
CLICK CHEMISTRY IN DRUG DISCOVERY
Fragment Based Drug Discovery and In Situ Click Chemistry
Fragment based drug discovery (FBDD) has emerged as a powerful complement to high throughput screening (HTS), leveraging small, low affinity fragments to explore chemical space efficiently [56,57]. In situ click chemistry, pioneered by Sharpless and Kolb in 2003 [57], extends FBDD by exploiting the protein target itself as the reaction template. Azide and alkyne functionalized fragments are incubated with the target enzyme; if two fragments bind in adjacent sub sites, the protein concentrates the reactive groups and catalyzes triazole formation. This thermodynamic selection process yields the most tightly binding combination of fragments, effectively allowing the enzyme to 'self assemble' its own inhibitor from a library of building blocks.
The in situ approach was elegantly demonstrated with acetylcholinesterase (AChE), where incubation with tacrine azide and phenylacetylene fragment libraries produced femtomolar inhibitors—among the most potent AChE inhibitors ever reported—without conducting conventional SAR campaigns [57]. Subsequent applications have extended the strategy to HIV protease, carbonic anhydrase, protein tyrosine phosphatases, and kinases. The method's intrinsic advantage is that it screens compound interactions directly in the context of the protein's active site, incorporating conformational selection and induced fit phenomena that solution phase screening cannot replicate.
Combinatorial Library Synthesis
The modularity and robustness of CuAAC have made it a pillar of combinatorial chemistry. Parallel click reactions between diverse azide and alkyne building blocks enable the rapid assembly of structurally diverse triazole libraries. Unlike classical combinatorial methods requiring protected amines and coupling agents, CuAAC is functional group tolerant, eliminating the need for protecting group strategies in many cases [16]. Libraries of thousands of compounds—pharmacologically relevant heterocycles, peptidomimetics, nucleoside analogues, and steroid derivatives—have been synthesised using automated CuAAC workflows [14].
A particularly productive application is the synthesis of triazole based bioisosteres of pharmacologically important scaffolds. Triazoles replace amide bonds, esters, and cis alkenes in drug leads, conferring metabolic stability and improved bioavailability. Anti infective triazole libraries have yielded potent inhibitors of Mycobacterium tuberculosis, Plasmodium falciparum, and Candida species [16]. In oncology, combinatorial click approaches have accelerated the identification of kinase inhibitors, HDAC inhibitors, and tubulin binding agents with sub nanomolar potency.
Activity Based Protein Profiling (ABPP)
Activity based protein profiling (ABPP) uses chemically reactive probes to covalently label the active sites of enzymes in a mechanism dependent fashion, enabling their detection, identification, and quantification in complex biological samples [44,45,46]. ABPP probes consist of three elements: a reactive warhead targeting specific enzyme classes (e.g., fluorophosphonate for serine hydrolases, vinyl sulfone for cysteine proteases), an affinity or reporter tag for enrichment and detection, and a linker connecting them. Click chemistry has transformed ABPP by permitting the design of minimally sized two part probes: a compact warhead bearing an azide or alkyne handle is administered to cells or whole organisms, and the reporter tag is appended post labelling via CuAAC or SPAAC [43,45].
This two step strategy offers several advantages over preformed probes: smaller probe size improves membrane permeability, the separation of warhead administration from reporter conjugation reduces non specific background, and the modular design allows probe structures to be optimised independently of the reporter. Cravatt and colleagues applied this strategy to map the human serine hydrolase and cysteine protease superfamilies globally in living cells, identifying previously uncharacterised enzymes and correlating their activity profiles with cancer phenotypes [44,45]. ABPP has since been extended to kinases (using acyl phosphates), phosphatases (using pervanadate based probes), glycosidases (using mechanism based sugar probes), and epigenetic enzymes including deacetylases and methyltransferases [46].
Competitive ABPP, in which small molecule drug candidates compete with the ABPP probe for active site occupancy, provides a quantitative, target agnostic measure of compound potency and selectivity across the entire enzyme proteome simultaneously [43]. This approach has revealed unexpected off targets for clinical drugs, guided the development of isoform selective inhibitors, and identified new druggable enzymes that had been invisible to traditional biochemical screening.
Covalent Drug Design and Click Chemistry
Covalent drugs, which form irreversible or slowly reversible bonds with their target proteins, have re emerged as a major therapeutic strategy following the clinical validation of drugs such as ibrutinib (BTK), osimertinib (EGFR), and sotorasib (KRAS G12C). Click chemistry intersects with covalent drug design at multiple points. First, the triazole motif itself can serve as a pharmacophore that positions reactive warheads relative to target residues [57]. Second, ABPP guided discovery of reactive cysteines—using iodoacetamide alkyne probes and quantitative mass spectrometry—has created a comprehensive map of covalently druggable residues across the human proteome [44,45,57].
Third, the SuFEx reaction—sulfur(VI) fluoride exchange, introduced by Sharpless and Dong as the 'next generation' click reaction—has opened entirely new avenues in covalent drug discovery [76]. Sulfonyl fluorides and fluorosulfates are bench stable electrophiles that react selectively with tyrosines, lysines, serines, histidines, and threonines in protein active sites, providing a broader coverage of covalently addressable residues than classical cysteine targeting strategies. SuFEx reagents have been applied in fragment based covalent screening, enabling the identification of selective covalent fragments against previously undruggable targets.
Nucleoside and Nucleotide Analogues
Nucleoside analogues represent a major class of antiviral and anticancer drugs. Click chemistry has dramatically expanded the diversity of accessible nucleoside scaffolds by enabling the modular conjugation of modified bases, sugars, and lipophilic tails [16]. CuAAC allows orthogonal functionalisation of nucleosides at the 2' , 3' , or 5' positions without protecting group manipulation, facilitating rapid SAR exploration of antiviral candidates against HIV, HCV, SARS CoV 2, and influenza. Triazole linked nucleoside dimers and oligonucleotide analogues display improved nuclease resistance and cellular uptake relative to natural phosphodiester linkages, and triazole linked DNA analogues have been demonstrated to support transcription and translation with high fidelity [16].
CLICK CHEMISTRY IN BIOCONJUGATION
Antibody Drug Conjugates (ADCs)
ADCs represent the most therapeutically impactful application of click bioconjugation. An ADC consists of a monoclonal antibody (mAb) conjugated via a chemical linker to a cytotoxic small molecule payload. The antibody provides tumour targeted delivery, while the payload delivers potent cell killing activity upon internalisation and linker cleavage [38,39]. As of 2024, more than 15 ADCs have received FDA approval, with a pipeline of over 100 in clinical trials. The chemistry of conjugation profoundly influences ADC pharmacokinetics, therapeutic index, homogeneity, and manufacturability.
Conventional ADC synthesis relies on non selective coupling of cytotoxic payloads to lysine ε amino groups or maleimide based thiol conjugation to inter chain cysteines exposed by disulfide reduction. Both approaches yield heterogeneous drug antibody ratio (DAR) distributions, compromising pharmacokinetic predictability [40]. Site specific conjugation, enabled by click chemistry, addresses this critical limitation. Three major click based strategies have emerged: (i) unnatural amino acid incorporation with amber suppression [41], (ii) enzyme catalyzed tag introduction (sortase, transglutaminase, formylglycine generating enzyme) followed by oxime or CuAAC conjugation [42], and (iii) SPAAC or IEDDA based conjugation to engineered reactive handles [39,42].
The axle for site specific ADC synthesis via unnatural amino acid incorporation was established by Schultz and colleagues [41], who demonstrated that p acetylphenylalanine (pAcF)—an unnatural amino acid with a ketone side chain—could be genetically encoded using an engineered pyrrolysyl tRNA synthetase/tRNA pair. The ketone serves as an oxime ligation handle for hydroxylamine functionalized payloads, yielding homogeneous DAR 2 ADCs with markedly improved therapeutic indices compared to statistical conjugates. The FDA approved ADC polatuzumab vedotin and others in the pipeline have adopted site specific conjugation strategies informed by click chemistry principles [38].
SPAAC based ADC synthesis employs DBCO functionalised payloads that react with azide bearing antibodies introduced enzymatically or through engineered glycosylation sites. This approach is operationally straightforward, copper free, and compatible with the scale and quality requirements of biopharmaceutical manufacturing [39]. Tetrazine TCO ligation has been explored for rapid dual click ADC assembly and pretargeted ADC strategies where the antibody and cytotoxin are assembled in vivo at the tumour site, minimising systemic toxicity [48,51].
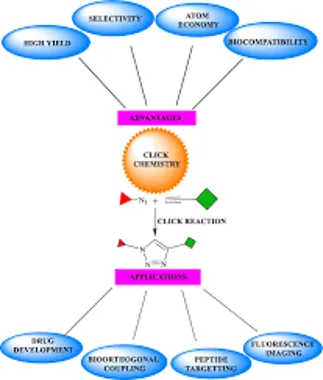
FIG.2 APPLICATION OF CLICK CHEMISTRY
Table 2. FDA Approved and Clinical Stage ADCs Utilizing Click Chemistry Conjugation Strategies
|
Agent |
Target |
Payload |
Conjugation |
Status / Indication |
|
Brentuximab vedotin |
CD30 |
MMAE |
Maleimide thiol (site specific variant) |
FDA approved; Hodgkin lymphoma, sALCL |
|
Trastuzumab emtansine |
HER2 |
DM1 |
Lysine SMCC (optimised click variant studied) |
FDA approved; HER2+ breast cancer |
|
Sacituzumab govitecan |
TROP 2 |
SN 38 |
Hydrolysable linker / oxime ligation |
FDA approved; TNBC, urothelial |
|
XDCX 001 (XDC) |
EGFR |
Auristatin |
SpAAC/bioorthogonal |
Phase I; solid tumours |
|
NC318 (click ADC) |
Siglec 15 |
MMAF |
SPAAC site specific |
Phase II; NSCLC |
|
Tz pretargeted RIT |
CD45 |
⁹⁰Y TCO |
IETDA (Tz/TCO) |
Phase I/II; AML conditioning |
ADC = antibody drug conjugate; MMAE = monomethyl auristatin E; DM1 = emtansine; SN 38 = irinotecan metabolite; MMAF = monomethyl auristatin F; SPAAC = strain promoted azide alkyne cycloaddition; Tz = tetrazine; TCO = trans cyclooctene; IETDA = inverse electron demand Diels Alder; NSCLC = non small cell lung cancer; AML = acute myeloid leukaemia; TNBC = triple negative breast cancer.
Site Specific Protein Modification
The ability to modify proteins at single, defined sites—with precise control over conjugation chemistry, stoichiometry, and spatial orientation—is a longstanding goal of chemical biology [63,64]. Click chemistry has provided the most versatile toolkit for this purpose. Four major strategies enable site selective protein functionalisation: (i) genetic encoding of unnatural amino acids bearing click handles (azides, alkynes, tetrazines, strained alkenes); (ii) enzymatic tag installation (ybbR tag/Sfp phosphopantetheinyl transferase, SNAP tag, Halo tag, SpyTag/SpyCatcher); (iii) N terminal selective condensation; and (iv) oxidative unnatural amino acid introduction [63,64].
The genetic code expansion approach, pioneered by Schultz and later extended by Chin's group [60], incorporates non canonical amino acids bearing azide (e.g., azidohomoalanine, p azidophenylalanine), alkyne (propargylglycine), cyclopropene, or tetrazine groups in response to amber stop codons. These bioorthogonal handles enable site specific CuAAC, SPAAC, or IEDDA conjugation in intact proteins, preserving tertiary structure and activity. The strategy has been applied to install fluorophores, PEG chains, lipid anchors, glycans, ubiquitin, and nucleic acids at predefined positions in proteins of pharmaceutical interest.
Enzyme mediated tagging strategies exploit the natural affinity and catalytic specificity of enzymes to install click handles at specific protein sequences. The formylglycine generating enzyme (FGE) converts cysteine to formylglycine (fGly) at a short consensus sequence, providing an aldehyde handle for oxime conjugation—a strategy commercialised as the aldehyde tag [42]. Sortase A mediates transpeptidation at LPXTG motifs, attaching azide or alkyne bearing peptides to the protein C terminus. Transglutaminase catalyzes the crosslinking of glutamine residues with lysine azide derivatives. All these strategies converge on click chemistry as the final conjugation step.
PEGylation and Polymer Bioconjugation
Polyethylene glycol (PEG) conjugation—PEGylation—is the dominant strategy for extending the plasma half life of therapeutic proteins and peptides, reducing immunogenicity, and improving solubility [70,71]. Conventional PEGylation via NHS ester or aldehyde chemistry is non selective, yielding heterogeneous positional isomers with variable biological activity. Site specific PEGylation via click chemistry has transformed this field by enabling precise, single site conjugation with defined PEG chain length and orientation [70].
CuAAC between alkyne PEG and azide protein is the most commonly used approach, although copper toxicity necessitates rigorous removal for clinical grade products [70]. SPAAC with DBCO PEG and azide proteins, and IEDDA with tetrazine PEG and TCO proteins, provide copper free alternatives with excellent efficiency. Click PEGylation has been applied to insulin, erythropoietin, interferon α, interleukins, and therapeutic antibody fragments (Fabs), consistently demonstrating superior pharmacokinetic profiles relative to conventional PEGylated analogues. The precision offered by click chemistry is particularly advantageous for peptide therapeutics, where PEG conjugation at a single defined position preserves receptor engagement while maximising exposure.
Glycan Engineering and Glycoprotein Synthesis
Glycoproteins—proteins decorated with complex oligosaccharide chains—mediate critical biological processes including cell adhesion, immune recognition, and protein folding. The synthesis and chemical modification of glycoproteins has historically been impeded by the structural complexity and lability of glycans. Click chemistry has provided elegant solutions to these challenges [22,30,34].
Metabolic glycan labelling exploits the cell's biosynthetic machinery to incorporate unnatural azide bearing monosaccharide analogues (N azidoacetylmannosamine, N azidoacetylgalactosamine, N azidoacetylglucosamine) into cell surface and secreted glycoproteins. Subsequent SPAAC or CuAAC conjugation installs reporters, drugs, or affinity handles on specific glycan types with exquisite selectivity—a strategy that has illuminated dynamic glycan remodelling during development [22], cancer progression [30], and viral infection. Bertozzi's laboratory applied this approach in living zebrafish embryos, imaging mucin type O linked glycans during gastrulation—the first metabolic labelling experiment performed in a vertebrate organism [22].
Chemoenzymatic glycan remodelling—glycan stripping followed by enzymatic re installation of defined sugar structures—combined with azide tagged sugars and subsequent SPAAC conjugation, has enabled the synthesis of homogeneous antibody glycoforms with defined effector function profiles. This is of immediate relevance to the engineering of next generation therapeutic mAbs where Fc glycosylation critically determines ADCC, CDC, and pharmacokinetic behaviour.
Nucleic Acid Bioconjugation
Click chemistry has also transformed nucleic acid bioconjugation. Azide and alkyne modified nucleotides are readily incorporated into DNA and RNA by polymerase chain reactions or in vitro transcription, enabling post synthesis click conjugation of fluorophores, biotin, proteins, nanoparticles, and lipid carriers [16]. Click nucleic acid conjugates are employed in: FISH probes for cytogenetic diagnostics, aptamer drug conjugates for targeted delivery, siRNA antibody conjugates, and nucleic acid nanotechnology for programmable drug delivery vehicles.
Triazole linked DNA oligonucleotides—in which click chemistry replaces the phosphodiester bond—have been shown to support faithful replication by DNA polymerases and transcription by RNA polymerases, demonstrating that the triazole bond can functionally substitute for natural phosphodiester linkages in hereditary information transfer. This extraordinary finding has implications for synthetic biology, molecular diagnostics, and the design of non natural nucleic acid therapeutics resistant to nuclease degradation.
CLICK CHEMISTRY IN IMAGING AND DIAGNOSTICS
Bioorthogonal Metabolic Labelling
The capacity to image specific biomolecules in their native biological context—without genetic manipulation of the target itself—represents one of the most transformative contributions of bioorthogonal click chemistry to cell and developmental biology [55,56,58]. The metabolic labelling strategy exploits the cell's biosynthetic pathways to incorporate bioorthogonal chemical reporters (azides, alkynes, tetrazines) into lipids, proteins, nucleic acids, and glycans, which are then detected by click mediated installation of fluorescent probes.
Laughlin et al. extended metabolic glycan labelling into living zebrafish embryos, using peracetylated N azidoacetylmannosamine (Ac₄ManNAz) to label sialic acids on mucin type glycoproteins and imaging them with DIFO biotin SPAAC [22]. The spatial and temporal resolution achieved revealed previously unappreciated patterns of glycoprotein trafficking during gastrulation. Analogous strategies have been applied to image de novo lipid synthesis (using alkyne fatty acids and CuAAC), protein synthesis (using azidohomoalanine/BONCAT), and DNA replication (using EdU—5 ethynyl 2' deoxyuridine—and CuAAC with Alexa Fluor azide dyes, now a clinical grade proliferation assay) [56].
PET and SPECT Radiolabelling
Positron emission tomography (PET) and single photon emission computed tomography (SPECT) require the attachment of short lived radioisotopes (¹⁸F, ⁶⁴Cu, ⁸⁹Zr, ⁹⁰Y, ¹¹¹In) to targeting vectors—typically antibodies, peptides, or small molecules—within timeframes dictated by isotope half lives and radiochemical stability. Click chemistry is ideally suited to these constraints, offering rapid, high yielding conjugations without the need for protecting groups or high temperature conditions [50,53].
CuAAC based ¹⁸F labelling using [¹⁸F]fluoroethyl azide and alkyne bearing peptides or proteins achieves radiochemical yields exceeding 90% in under 30 minutes—well within the 110 minute half life of ¹⁸F [50]. IEDDA tetrazine TCO chemistry, with its exceptionally fast kinetics, is particularly advantageous for pretargeted immunoPET: a TCO antibody is administered and allowed to accumulate at the tumour over 24–48 hours, and a small tetrazine ¹⁸F or tetrazine ⁶⁴Cu agent is subsequently injected and 'clicks' onto the accumulated antibody in seconds, providing near instantaneous target engagement with low background [51,52].
Zeglis, Lewis, and colleagues demonstrated pretargeted PET imaging of colorectal cancer xenografts in mice using a huA33 TCO antibody and a ⁶⁴Cu NOTA tetrazine radioligand, achieving tumour to blood ratios far superior to directly radiolabelled antibody constructs [51]. Clinical translation of pretargeted radioimmunotherapy (PRIT) using ⁹⁰Y tetrazine and anti CD45 TCO antibodies for AML conditioning prior to stem cell transplantation is actively under investigation in Phase I/II trials [52].
Fluorescence Imaging and Super Resolution Microscopy
Click chemistry mediated fluorophore conjugation has become the standard for specific, low background fluorescent labelling of biomolecules in cells and tissues [27,56]. The small size of azide and alkyne handles—compared to fluorescent protein tags or antibodies—minimises steric perturbation and enables labelling in sterically challenging environments such as the nuclear pore complex, ribosome, and viral capsid. IEDDA based fluorophore conjugation, exploiting the ultra fast kinetics of tetrazine TCO ligation, enables single step, wash free live cell imaging at nanomolar probe concentrations, dramatically improving signal to noise ratios.
In super resolution microscopy—STORM, PALM, STED, and PAINT—click conjugation of small organic fluorophores provides the photophysical properties (high photon output, controlled blinking, small labelling radius) required for nanometre scale structural resolution, surpassing what is achievable with fluorescent protein fusions [56]. DNA PAINT—which uses complementary azide/alkyne functionalised DNA strands transiently binding to target localised handles—leverages click chemistry for probe immobilisation and has resolved protein assemblies at 5–10 nm resolution in fixed cells.
Table 3. Comparative Kinetics and Bioorthogonality of Major Click Reaction Pairs for Biological Applications
|
Click Pair |
k₂ (M⁻¹s⁻¹) |
Bioorthogonal |
Cu Free |
Biological Use Cases |
|
CuAAC (azide/ alkyne) |
10–100 |
Partial (Cu toxic) |
No |
In vitro, ex vivo proteomics, materials |
|
SPAAC (DIBO/DIFO) |
0.1–1.0 |
Yes |
Yes |
Live cell glycan labelling, protein PEGylation |
|
DBCO azide |
0.3–0.97 |
Yes |
Yes |
In vivo tumour imaging, prodrug activation |
|
Tetrazine/TCO (IEDDA) |
10³–10⁶ |
Yes |
Yes |
Pretargeted PET, rapid protein tagging |
|
Tetrazine/norbornene |
~1 |
Yes |
Yes |
Slow but stable; hydrogel crosslinking |
|
Staudinger ligation |
~0.002 |
Yes |
Yes |
Glycan imaging; slow kinetics limit in vivo |
|
Thiol ene (photoclick) |
~10³ (UV) |
Partial |
Yes |
Surface patterning, hydrogel fabrication |
k₂ = second order rate constant; SPAAC = strain promoted azide alkyne cycloaddition; DIBO/DIFO = cyclooctyne derivatives; DBCO = dibenzocyclooctyne; IEDDA = inverse electron demand Diels Alder; TCO = trans cyclooctene; Tz = tetrazine; BONCAT = bio
BIOMATERIALS
Polymer Based Nanocarriers
Click chemistry has been extensively exploited in the construction of polymer based drug delivery systems, including polymeric nanoparticles, dendrimers, micelles, and vesicles. The orthogonality and efficiency of CuAAC, thiol ene, and IEDDA reactions enable precise control over polymer architecture, surface functionalisation density, targeting ligand orientation, and drug loading [69,72]. PLGA, PEG PLGA, and poly(lactide co glycolide) nanoparticles functionalised with folate alkyne or transferrin azide via CuAAC display enhanced tumour uptake relative to non targeted counterparts in both in vitro and animal models [54,69].
SPAAC and IEDDA chemistry are preferred when copper free in vivo assembly is required. In a landmark study, Koo et al. demonstrated that in vivo SPAAC between azide functionalised nanoparticles and DBCO antibody fragments pre localised at tumour vasculature enabled targeted nanoparticle accumulation in mouse xenograft models with tumour to background ratios far exceeding passively targeted controls [54]. This 'in vivo click assembly' strategy decouples nanoparticle preparation from targeting ligand conjugation, providing fresh, stable targeting moieties at the tumour surface.
Hydrogels and Controlled Release
Hydrogels—crosslinked hydrophilic polymer networks—serve as scaffolds for tissue engineering, cell encapsulation, wound healing, and sustained drug release. Click crosslinking, particularly thiol ene and IEDDA reactions, has enabled the design of hydrogels with precise control over network topology, mechanical properties, degradation kinetics, and cell adhesion peptide presentation [36,55,68]. Photo initiated thiol ene click reactions enable spatiotemporal control over hydrogel gelation and property modulation, enabling user defined micropattern formation relevant to guided cell migration and tissue morphogenesis.
Tetrazine TCO IEDDA crosslinked hydrogels form within seconds upon mixing of tetrazine polymer and TCO polymer precursors under physiological conditions, without UV irradiation or added catalyst—a major advantage for cell encapsulation and in situ gelation at injection sites [55]. These hydrogels have been used for the sustained local delivery of growth factors, antibiotics, chemotherapeutics, and nucleic acid therapeutics. The reversibility of some click crosslinks (particularly Diels Alder adducts at elevated temperatures) has been exploited to engineer shape memory and self healing hydrogels, and for triggered drug release using thermal or chemical stimuli.
Surface Functionalisation
Functionalisation of implant surfaces, biosensor platforms, microfluidic devices, and diagnostic arrays with biomolecules requires chemistries that are efficient, orientationally controlled, and stable under operational conditions. CuAAC and thiol ene click reactions provide ideal surface chemistry by enabling high density, covalently stable attachment of proteins, nucleic acids, carbohydrates, and small molecules to azide , alkyne , or thiol bearing surfaces [68]. Click functionalised surfaces have been used in the development of ELISA platforms with enhanced sensitivity, protein microarrays for serum biomarker profiling, implantable glucose sensors with anti fouling PEG coatings, and bactericidal titanium implants decorated with antimicrobial peptides.
EMERGING FRONTIERS IN CLICK CHEMISTRY
Sulfur(VI) Fluoride Exchange (SuFEx)
SuFEx chemistry, unveiled by Sharpless and Dong in 2014 [76], represents the most significant expansion of the click chemistry toolkit in the past decade. SuFEx reactions involve the exchange of fluoride at sulfur(VI) centres—sulfonyl fluorides (SO₂F), fluorosulfates (OSO₂F), and related species—with oxygen, nitrogen, or sulfur nucleophiles under mild conditions catalysed by organic bases. The reaction is exceptionally functional group tolerant, high yielding, and scalable, making it ideal for industrial synthesis of polyurethanes, polysulfonates, and polysulfonamides from simple bifunctional SuFEx building blocks.
In drug discovery, SuFEx's most transformative impact is in covalent fragment screening and targeted covalent inhibitor design. Aryl sulfonyl fluorides react selectively with tyrosine, lysine, serine, and histidine residues in protein active sites, expanding the repertoire of covalently targetable residues well beyond the classical cysteine reactive warheads. The Sharpless group applied SuFEx to build a library of 1,4 bis(fluorosulfonyl)benzene linkers for modular inhibitor synthesis, demonstrating picomolar inhibitors of carbonic anhydrase assembled in the enzyme's active site by double SuFEx reaction [76]. ¹⁸F labelled sulfonyl fluorides have been explored as PET tracers for covalent protein imaging, and fluorosulfate isosteres of phosphate bioisosteres are increasingly exploited in medicinal chemistry [77,78].
Photoclick Chemistry
Photoclick chemistry uses light to trigger or accelerate click reactions with spatial and temporal precision unattainable by thermally driven reactions. The most important photoclick reactions are: (i) tetrazole alkene photocycloaddition via nitrile imine intermediates [11], (ii) photo induced thiol ene reactions, and (iii) photogenerated cyclopentadienyl species for IEDDA reactions. Tetrazole photoclick, introduced by Lin and Turro, proceeds upon UV irradiation (λ ≈ 302–365 nm) of diaryltetrazoles that photolyse to reactive nitrile imines, which undergo [3+2] cycloaddition with alkenes or activated alkynes to yield fluorescent pyrazoline products—providing a self reporting labelling strategy [11].
The spatiotemporal control offered by photoclick chemistry enables unprecedented applications: photopatterning of hydrogel surfaces for directed cell migration, light triggered drug release from prodrug conjugates, pulse labelling of newly synthesised proteins in defined subcellular compartments, and photo directed assembly of nanostructures. Two photon photoclick reactions extend this control to three dimensional spatial resolution within living tissues, opening possibilities for optogenetically controlled bioconjugation in vivo [72].
Click to Release Chemistry
A conceptually elegant extension of click chemistry is 'click to release'—the use of a bioorthogonal reaction not to form a stable product but to trigger the elimination of a masked molecule. Versteegen, Rossin, and Robillard demonstrated that tetrazine reacts with TCO carbamate prodrugs via IEDDA followed by spontaneous 1,6 elimination to release free primary amines or hydroxyl groups—activating prodrugs quantitatively in aqueous media at ambient temperature [48,49]. This platform has been applied to activate doxorubicin, paclitaxel, duocarmycin, and antibiotics from TCO prodrug conjugates in vivo using systemically administered tetrazine [48,49].
The click to release strategy decouples drug localisation from drug activation, enabling a pharmacological separation between the toxophore delivery step (via the antibody or nanoparticle carrier) and the chemical activation step (via the click reagent). This approach has been applied in pretargeted ADC strategies and as a 'chemical antibody' mediated drug activation system, with Phase I/II clinical trials initiated for AML treatment using anti CD45 TCO antibody pretargeting followed by ⁹⁰Y DOTA tetrazine administration [52].
Dual Click and Multi Orthogonal Strategies
The availability of multiple mutually orthogonal click reactions has enabled the simultaneous labelling of two or more biomolecules in the same sample or organism [55,56]. True multi orthogonality requires that each click pair reacts exclusively with its counterpart without cross reactivity. CuAAC (azide/alkyne), SPAAC (azide/cyclooctyne), and IEDDA (tetrazine/TCO) are mutually orthogonal when the cyclooctyne and TCO substrates are sufficiently differentiated, enabling dual metabolic labelling of glycans and proteins, or glycans and lipids, with distinct fluorophores in a single experiment. In materials science, dual click sequential crosslinking—first IEDDA to establish network topology, then UV thiol ene to tune mechanical properties—creates programmable biomimetic scaffolds with spatially defined mechanical gradients.
Split and pool multi orthogonal click synthesis has been used to construct peptide heterocycle polymer hybrid materials with unprecedented structural complexity from simple building blocks, illustrating the combinatorial power that multi orthogonal click chemistry can deliver. Future developments in this area will likely incorporate bio catalytic click reactions—enzyme catalyzed, genetically programmable bioconjugation reactions that achieve the speed and selectivity of biochemical modifications within synthetic chemistry frameworks.
TRANSLATIONAL CHALLENGES AND CLINICAL PERSPECTIVES
Copper Toxicity and Mitigation Strategies
Despite CuAAC's unparalleled utility in vitro and in materials synthesis, copper's cytotoxicity presents a fundamental challenge for in cellulo and in vivo applications. Copper mediated generation of reactive oxygen species causes DNA strand breaks, protein carbonylation, and lipid peroxidation at concentrations (>10 µM) often required for efficient CuAAC in complex biological matrices [21]. Copper chelating ligands—THPTA, BTTES, BTTAA—substantially reduce required copper concentrations (to 10–50 µM) by increasing catalytic efficiency and scavenging oxidative byproducts, enabling live cell CuAAC labelling with acceptable cytotoxicity [21]. Sodium ascorbate, the reductant used to generate Cu(I) in situ, also serves as an antioxidant.
For therapeutic applications where copper residuals in biopharmaceuticals must meet regulatory limits (ICH Q3D: ≤250 µg/day oral intake), extensive downstream purification is required following CuAAC based conjugation. This adds process complexity and cost to ADC and PEGylated protein manufacture. Copper free SPAAC and IEDDA reactions entirely circumvent this issue and are increasingly preferred for clinical grade bioconjugation. Nevertheless, CuAAC remains indispensable for chemical library synthesis, polymer chemistry, and ex vivo biological studies where copper removal is straightforward.
Pharmacokinetic Properties of Triazole Containing Drugs
The 1,4 disubstituted 1,2,3 triazole ring's physicochemical properties—high polarity (cLogP reduced by ~0.5 per triazole), resistance to oxidative metabolism, and potential for dipole directed interactions with target residues—generally translate into favourable pharmacokinetic profiles [14,16]. However, highly polar triazole containing compounds may exhibit limited membrane permeability and oral bioavailability, particularly those derived from fragment linking strategies where two polar fragment pharmacophores are bridged by the triazole linker. Strategic placement of the triazole at sites not critical for membrane permeation, or replacement with lipophilic analogues (e.g., thio triazoles or methyl triazoles), can mitigate these effects.
Metabolic stability of the triazole ring itself is generally excellent; CYP450 mediated N dealkylation of N substituted triazoles can occur but is significantly slower than for imidazoles or pyrazoles. Triazole antifungals (fluconazole, voriconazole, posaconazole) demonstrate that the triazole scaffold can achieve excellent oral bioavailability when the surrounding molecule is appropriately designed. As click chemistry derived drug candidates progress through preclinical development, comprehensive metabolite identification studies using ¹⁴C triazole labelled probes and liver microsomal assays are recommended to identify soft spots and guide structural optimisation.
Linker Stability in ADCs
Linker design is arguably the most critical determinant of ADC therapeutic index. A linker must be sufficiently stable in circulation to prevent premature payload release (causing systemic toxicity) while enabling rapid, complete release upon internalisation into the target cancer cell's lysosomal compartment. Click chemistry based linkers—particularly maleimide thiol, oxime, and hydrazone linkers—display distinct stability profiles that must be carefully matched to the pharmacokinetic requirements of the specific ADC [38,39].
Maleimide thiol adducts, while kinetically stable, can undergo retro Michael addition (maleimide exchange) in plasma, particularly in the first 24 hours post administration, leading to premature payload transfer to serum albumin and other thiol bearing proteins [39,40]. Hydrolysis of the succinimide ring—either spontaneously or catalysed by hydrophobic microenvironments—generates an open chain thioether that is substantially more stable. Careful pH adjustment (pH 8.0–9.5) during ADC synthesis promotes succinimide hydrolysis, yielding more stable conjugates, a strategy incorporated into the manufacture of several approved ADCs [39]. Alternative thiol click reactions—using self hydrolyzing or sterically hindered maleimides—are actively investigated for next generation ADCs.
Immunogenicity and Biocompatibility of Click Reagents
Azides, alkynes, cyclooctynes, tetrazines, and TCO are 'non biological' functional groups absent from mammalian biochemistry—the very property that confers their bioorthogonality. This also raises questions about their immunogenicity and biocompatibility at the concentrations and exposure durations relevant to therapeutic use. Immunotoxicology studies on DBCO functionalised ADCs and TCO bearing antibodies have generally not revealed hapten specific immune responses or complement activation, though systematic studies are limited and represent an active area of regulatory science [56].
Acute cytotoxicity studies in rodent models using clinically relevant doses of DBCO, BCN, and tetrazine reagents have shown acceptable tolerability, supporting the safety case for click bioorthogonal bioconjugates. The FDA's Guidance for Industry on ADC development does not specifically address bioorthogonal linker chemistry, reflecting the novelty of the field; however, the established regulatory frameworks for conventional ADC linkers and site specific conjugation strategies provide a robust precedent for seeking approval of click chemistry derived conjugates.
FUTURE PERSPECTIVES
The trajectory of click chemistry points toward increasingly sophisticated, biologically integrated applications that blur the boundaries between synthetic chemistry and living systems. Several themes are likely to define the next decade of the field.
First, artificial intelligence and machine learning are poised to accelerate click chemistry guided drug discovery. Transformer based generative models trained on click accessible chemical spaces can propose novel azide alkyne fragment pairs with predicted target affinity, and reinforcement learning algorithms can optimise synthetic routes that maximise click steps for efficiency. The integration of ABPP proteomic data with machine learning target prediction will identify new covalently addressable drug targets with unprecedented speed [73].
Second, the development of genetically encodable bioorthogonal reactions—click reactions catalysed by engineered or evolved enzymes within living cells—could eliminate the need for exogenous chemical reagents entirely for intracellular bioconjugation applications. Directed evolution of pyrrolysyl tRNA synthetases to accept increasingly diverse unnatural amino acids bearing bioorthogonal handles is expanding the genetic code repertoire and enabling multi site, multi click labelling from a single genetic construct.
Third, the convergence of click chemistry with CRISPR based genome editing, RNA therapeutics, and cell and gene therapy presents enormous opportunities. Click mediated functionalisation of lipid nanoparticles (LNPs) for mRNA and siRNA delivery—attaching targeting ligands, endosomal escape peptides, and diagnostic reporters to LNP surfaces via IEDDA or SPAAC—may dramatically improve the tissue specificity and potency of next generation RNA medicines. Click modified guide RNAs and base editors are being explored for improved CRISPR delivery and activity.
Fourth, in vivo click chemistry will increasingly transition from proof of concept demonstrations to clinical tools. Pretargeted PET imaging using tetrazine TCO ligation has already entered clinical trials, and pretargeted radioimmunotherapy using click to release strategies is advancing through Phase I/II evaluation. As pharmacokinetic data from early clinical trials accumulate, the parameters governing optimal TCO antibody dosing, tetrazine pharmacokinetics, and bioorthogonal reaction efficiency in human tumour microenvironments will be defined, enabling rational design of subsequent clinical programmes.
Finally, sustainability and green chemistry principles are increasingly being applied to click reaction design. Mechanochemical CuAAC—performing click reactions by ball milling without solvent—achieves quantitative yields with minimal waste and is amenable to pharmaceutical manufacturing at scale. Photocatalytic CuAAC using visible light copper photocatalysts operates at ambient temperature without stoichiometric reductants, further reducing the environmental footprint of click based synthesis.
CONCLUSION
Click chemistry has traversed a remarkable trajectory—from an elegant synthetic philosophy articulated in 2001 to a Nobel Prize winning technology platform integral to modern pharmaceutical science, chemical biology, and materials engineering. The convergence of CuAAC, SPAAC, IEDDA, Staudinger ligation, oxime/hydrazone condensation, thiol ene addition, and the newest entrant SuFEx has created a versatile, modular toolkit capable of addressing virtually any bioconjugation or synthetic challenge in drug discovery.
In drug discovery, click chemistry's impact spans target identification (ABPP), hit finding (in situ click, FBDD), lead optimisation (triazole bioisosteres, covalent drug design), and clinical translation (ADCs, PEGylated biologics, imaging agents). The precision of site specific click bioconjugation has transformed ADC manufacturing, enabling homogeneous conjugates with superior therapeutic indices that are now routinely entering clinical trials. Bioorthogonal click chemistry has opened the living cell and the living animal as laboratories for precise chemical intervention, enabling real time imaging of molecular events, metabolically labelled biosynthetic processes, and light directed in vivo drug activation.
Challenges remain—copper toxicity, linker stability, pharmacokinetic liabilities of triazole rich molecules, and regulatory uncertainty around novel bioorthogonal linker chemistries—but each is being systematically addressed by the field's ingenuity. The combination of click chemistry's modularity with artificial intelligence, genetic code expansion, RNA therapeutics, and sustainable manufacturing practice suggests that the field is still at an early stage of its impact on human health. Click chemistry, true to its founding promise, continues to find new, near perfect reactions—and new, near limitless applications—in the service of medicine and science.
REFERENCES



Nitish Kumar, Jibin Mathew, Dipanwita Pal, Click Chemistry as a Versatile Platform in Modern Drug Discovery and Bioconjugation, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5511-5532. https://doi.org/10.5281/zenodo.20790801
 10.5281/zenodo.20790801
10.5281/zenodo.20790801