
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shree Gulabrao Deokar College Of Pharmacy, Jalgaon
Cancer remains a leading cause of mortality worldwide, with drug resistance representing the primary barrier to sustained therapeutic success. Drug resistance may be classified as intrinsic (pre-existing) or acquired (developing under therapeutic pressure .and arises through diverse molecular, genetic, epigenetic, and microenvironmental mechanisms. This review systematically examines the classification, molecular mechanisms, clinical significance, and strategies to overcome drug resistance in cancer, with a focused case study of cisplatin resistance in ovarian cancer.Key resistance mechanisms include enhanced drug efflux via ABC transporters (particularly P-glycoprotein), mutations in drug targets such as receptor tyrosine kinases, upregulation of DNA repair pathways (NER, HR), impaired apoptosis through Bcl-2 and p53 dysregulation, and epigenetic alterations including DNA methylation and histone modifications. The tumor microenvironment contributes through hypoxia-driven HIF-1? activation, immunosuppressive cellular networks (MDSCs, TAMs, Tregs), and stromal interactions. Cancer stem cells further drive multidrug resistance via quiescence, superior drug efflux, and activation of Wnt/?-catenin, Notch, and PI3K/Akt pathways.Strategies to overcome resistance include combination chemotherapy, precision medicine guided by genomic profiling, nanotechnology-based drug delivery, CRISPR-Cas9 gene editing, immune checkpoint inhibition, CAR-T cell therapy, and epigenetic agents. Future directions encompass artificial intelligence for resistance prediction and liquid biopsy-based real-time monitoring. Overcoming drug resistance demands mechanistic insight, innovative multi-modal therapies, and precision oncology to improve patient outcomes globally
Cancer comprises a heterogeneous group of diseases characterized by uncontrolled proliferation and dissemination of abnormal cells.If the spread of cancer cells this stage is known as metastasis is not controlled, it can result in death. Cancer is caused by many external factors (tobacco, chemicals, radiation and infectious organisms) as well as some internal factors (inherited mutations, hormones, immune conditions and random mutations). The causes of cancer are diverse,complex and only partially understood. Many things are known to increase the risk of cancer, including dietary factors, certain infections, lack of physical activity,obesity and environmental pollutants . These factors may act together to initiate or promote carcinogenesis in human body and thus cancer is leading cause of death. Cancer has become one of the causes of death in India. It is estimated that there are nearly 2 to 2.5 million cancer cases at any given point of time. Over 7 lakhs new cases and 3 lakhs Deaths occur annually due to cancer.Nearly 15 lakh patients require facilities for diagnosis,treatment and follow up at a given time[1]. Cancer therapy has progressed remarkably over the past decades, yet the development of drug resistance continues to limit the long-term success of treatment. Many patients initially respond well to chemotherapy, targeted therapy, hormonal therapy, or immunotherapy, but tumors often adapt and become unresponsive, leading to disease recurrence and treatment failure . Drug resistance may be present from the beginning (intrinsic resistance) or may arise after repeated exposure to anticancer agents (acquired resistance). This ability of cancer cells to survive therapeutic pressure reflects a wide range of molecular, genetic, and environmental adaptations . A key contributor to resistance is the increased activity of efflux transporters, particularly ATP-binding cassette (ABC) proteins, which actively pump anticancer drugs out of tumor cells and reduce their intracellular concentration. Mutations or structural changes in drug targets, such as receptor tyrosine kinases, also reduce the effectiveness of targeted therapies . Epigenetic alterations—including DNA methylation changes, chromatin remodeling, and microRNA regulation—further influence cellular behavior and contribute to therapy evasion . In addition to these intrinsic cellular mechanisms, external factors within the tumor microenvironment, such as hypoxia, stromal interactions, and altered metabolic conditions, create protective niches that diminish the sensitivity of cancer cells to treatment . The presence of cancer stem cells (CSCs), a small population of highly resilient cells capable of self-renewal, also plays a central role in multidrug resistance and tumor regrowth [2, 3].
1.1 Types of Cancer:
Cancers are broadly classified into five categories based on tissue of origin:
• Carcinomas (80–90% of all cases): Originate from epithelial cells lining organs and skin (e.g., lung, breast, colon, prostate cancers).
• Sarcomas (~1%): Arise from connective tissues—bone, cartilage, fat, muscle (e.g., osteosarcoma, liposarcoma).
• Leukemias: Blood-forming cell malignancies producing dysfunctional white blood cells; most common pediatric cancer (e.g., ALL, AML, CLL).
• Lymphomas: Lymphatic system cancers from abnormal lymphocyte proliferation; two main types—Hodgkin and non-Hodgkin lymphoma.
• Central Nervous System Cancers: Arising from brain or spinal cord tissues.
Organ-Based Classification
Common organ-specific cancers include colorectal, lung, liver, stomach, cervical, bladder, esophageal, nasopharyngeal, and non-Hodgkin lymphoma. Each has distinct risk factors, screening protocols, and treatment approaches [4].
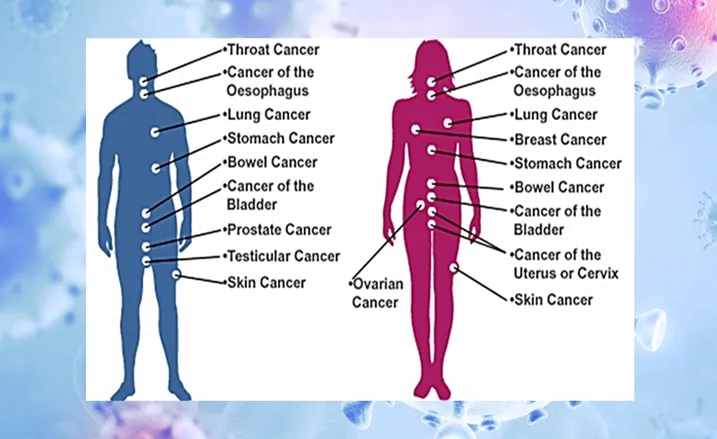
Figure 1.Types of Cancer
1.2 Symptoms Of Cancer General symptoms include fatigue, unexplained weight loss, night fever, loss of appetite, persistent pain, and abnormal skin changes. Organ-specific symptoms include blood in urine or stool, coughing up blood, and new lumps or masses.

Figure 2. Symptoms Of Cancer
1.3 Etiology & Pathophysiology
1.3.1 Etiology
Cancer cells display significant genetic instability and accelerated growth, allowing them to swiftly adjust to treatment-related pressures. Tumors are diverse, composed of different cell groups that exhibit varying responses to therapy. Certain cells may naturally have resistance mechanisms, resulting in intrinsic resistance.
Under therapeutic pressure, vulnerable cells are destroyed while resistant clones endure and multiply, leading to acquired resistance. Moreover, cancer stem cells play a role in resistance because of their dormant nature, effective DNA repair systems, and increased ability to survive
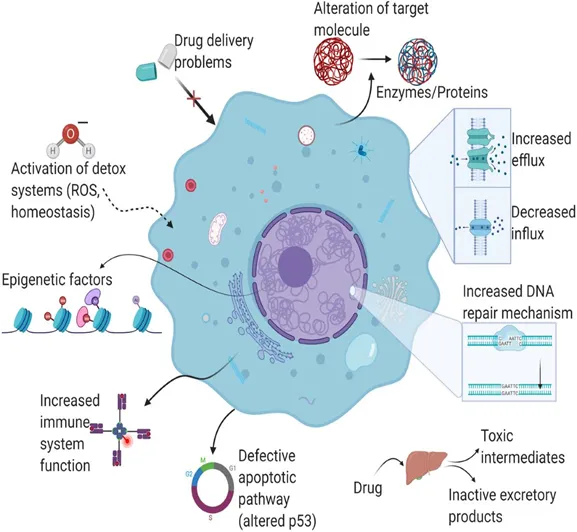
Figure 3. Etiology & Pathophysiology
1.3.2 Pathophysiology:
The pathophysiology of drug resistance in cancer reflects a complex set of adaptive responses that help tumor cells survive therapeutic stress. One of the most prominent mechanisms involves the increased activity of ATP-binding cassette (ABC) transporters, such as P-glycoprotein, which actively expel anticancer drugs from the intracellular space, lowering drug levels to sub-therapeutic concentrations. Resistance also arises from mutations within critical drug-interacting proteins, including receptor tyrosine kinases, topoisomerases, and cytoskeletal components, resulting in diminished drug binding and loss of treatment efficacy. Tumor cells may additionally upregulate DNA repair pathways such as homologous recombination and nucleotide excision repair, allowing them to quickly correct therapy-induced DNA damage and tolerate DNA-targeting agents[5]. Epigenetic changes further contribute to resistance. Alterations in DNA methylation, histone structure, and microRNA expression can shift gene regulation in favor of cell survival, reduced apoptosis, and adaptation to ongoing drug exposure. Many cancer cells enhance survival by activating compensatory signaling networks—such as PI3K/Akt, MAPK/ERK, and NF-κB—that maintain proliferation even when primary pathways are inhibited by treatment. The tumor microenvironment (TME) also plays an essential role; conditions such as low oxygen levels, acidic pH, cytokine release, and supportive stromal interactions create a physical and biochemical environment that protects tumor cells and decreases drug responsiveness . A distinct population known as cancer stem cells (CSCs) adds another layer to the resistance process. CSCs possess strong DNA repair capacity, low proliferative rates, and high survival potential, making them significantly more tolerant to chemotherapy and targeted therapy. Their persistence promotes tumor regrowth and is closely associated with multidrug-resistant behavior[6].
1.4 Chemotherapy and Treatment Ineffectiveness
Chemotherapy continues to be fundamental in cancer therapy, focusing on fast-dividing cells. Nonetheless, its efficacy is frequently restricted by the emergence of chemoresistance.
Chemotherapy failure (chemoresistance) occurs when tumors cease responding to drugs. It manifests as two subtypes:
• Intrinsic Resistance: Pre-existing resistance mechanisms render treatment ineffective from the outset.
• Acquired Resistance: Cancer cells develop resistance during therapy through adaptive molecular changes.
Key mechanisms behind chemotherapy failure include:
• Enhanced DNA Repair: Resistant cells rapidly repair chemotherapy-induced DNA damage.
• Drug Efflux Pumps: P-glycoprotein and other ABC transporters actively expel drugs from cells, reducing intracellular concentrations.
• Target Alteration: Mutations or downregulation of drug-binding proteins render drugs ineffective.
• Drug Inactivation: Enzymatic degradation or modification neutralizes chemotherapeutic agents.
• Gene Amplification: Overproduction of resistance-mediating proteins allows cancer cells to bypass drug effects.
3 Overcoming Chemotherapy Failure Strategies
include combination therapy (multiple mechanisms of action reduce resistance probability), targeted therapies and immunotherapy, dose/schedule modification, and genomic sequencing to guide personalized treatment selection. Mechanistically, resistance occurs through various pathways such as increased drug efflux, improved DNA repair, mutations in drug targets, and avoiding programmed cell death.[7].
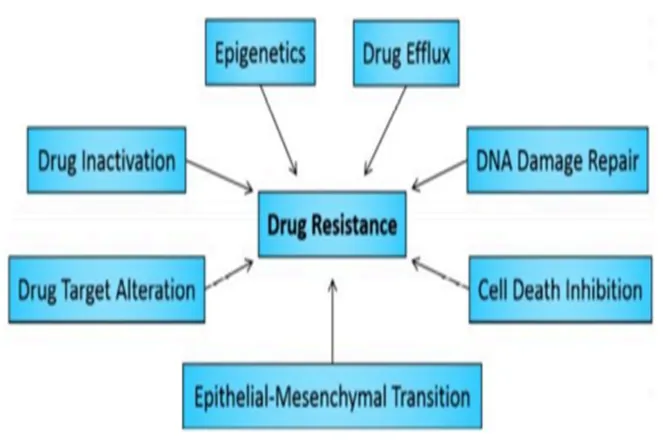
Figure 4. Categories of mechanisms that can enable or promote direct or indirect drug resistance in human cancer cells.
1.5 Importance in clinical oncology
Grasping drug resistance is crucial in contemporary oncology for multiple reasons:
• Facilitates the creation of targeted medicine strategies.
• Enhances the efficacy of treatment and overall survival rates.
• Aids in the advancement of clinical trial methodologies.
• Enables collaborative planning for diverse treatment approaches.
• Improves the quality of life for patients.
• Encourages strategies for early detection and prevention.
3 Clinical categorization of drug resistance
Drug resistance can be broadly divided into two main types: intrinsic resistance, which occurs naturally before treatment starts, and acquired resistance, which develops during or after treatment. Intrinsic resistance indicates that the treatment is not effective right from the beginning, as resistance mechanisms are already present. Acquired resistance, on the other hand, appears after treatment begins, suggesting that the treatment initially worked but later failed due to the development of resistance. This loss of effectiveness significantly impacts the chances of achieving complete remission.
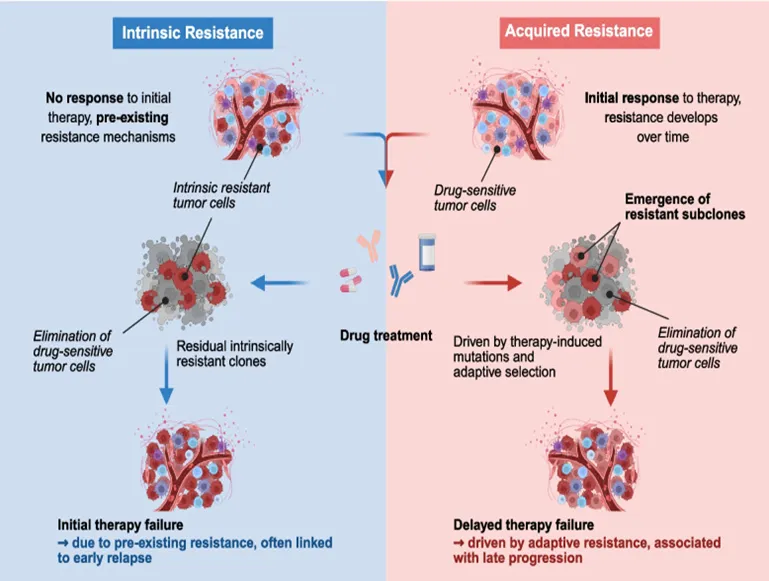
Figure 5. IntrinsicResistance & Acquired Resistance
3.1 Intrinsic resistance
Intrinsic resistance happens when patients fail to respond to treatment even with immune system participation. This could stem from an insufficient immune reaction to tumors, observed in those with compromised immune systems (for instance, HIV patients or organ transplant recipients) or elderly individuals whose T-cell variety is reduced. These individuals face an increased risk of reactivating infections or viruses. Tumors exhibiting a limited number of identifiable antigens (self-antigens compared to neo-antigens) hinder immune recognition, especially in cancers with low mutations, which makes it more challenging for the immune system to attack them. Moreover, tumors might be deficient in immune cells, indicating an unsuitable setting for immune function. Although T-cell responses are triggered, the tumor microenvironment may hinder their effectiveness through mechanisms such as PD-L1 expression and suppressive factors like TGF-β, IL-10, and IDO, which diminish T-cell activity and promote immune suppression through different cell types. Crucially, the existence of T-cells does not ensure successful targeting of the tumor. Intrinsic resistance also influences targeted treatments; for example, although BRAF inhibitors are effective in melanoma, colorectal cancers with the same mutation frequently fail to respond because of EGFR expression. Grasping these mechanisms is crucial for defeating inherent resistance.
3.2 Acquired Resistance
Naturally acquired resistance is specific to immunotherapy, marked by a decreased treatment response that arises from ongoing immune pressure rather than from cancer therapies. Patients often show signs of an active immune response in peripheral blood or tumor tissue, yet do not benefit from immune-modulating treatments. Current evidence on changes in T-cell activation or migration in this context is limited. Resistance mainly manifests through mechanisms that impair T-cell activity in the tumor microenvironment, involving inhibitory feedback mechanisms with various checkpoint molecules like LAG-3, TIM-3, and BTLA. For instance, engaged tumor-infiltrating effector T-cells may produce IFN-γ, inducing PD-L1 expression on tumor cells, which then interacts with PD-1, limiting T-cell function. Additionally, the immune response may select for tumor subpopulations lacking MHC class I or defective antigen processing, camouflaging the tumor from immune detection. This immune evasion can also occur as heterogeneous tumors yield subclones that lack certain antigens, known as immune-editing. Evidence for immune-editing exists in mouse models, but findings suggest that antigen loss might be less impactful when IFN-γ and TNF-α from cytotoxic T-cells disrupt tumor stroma. Human data remains scarce, though advances in identifying T-cell responses to (mutant) antigens may enhance understanding [8].
3.3 Multi drug resistance
In the context of cancer treatment, multi-drug resistance (MDR) refers to the capacity of cancer cells to survive treatment with multiple anticancer drugs (30), similar to the concept commonly used in antibiotic treatment. Cancer patients can receive two types of treatment: local and systemic. Radiation and surgery are categorized as local treatments, while chemotherapy, hormone therapy, and targeted therapy are considered systemic treatments. Systemic treatments are especially effective against metastatic or advanced-stage cancers. Increasing evidence suggests that MDR is mainly due to the enhanced efflux of chemotherapeutic drugs, which reduces their uptake by cancer cells. The mechanism of MDR may also involve the expulsion of drugs from the cells. MDR can develop due to oncogene mutations, changes in the tumor microenvironment (TME), tumor heterogeneity, mutations at the target sites, or epigenetic modifications. [9].

Figure 6. Compare Intrinsic& Acquired Resistance.
4 Molecular Mechanisms of Drug Resistance
4.1 Presence of Efflux Pumps
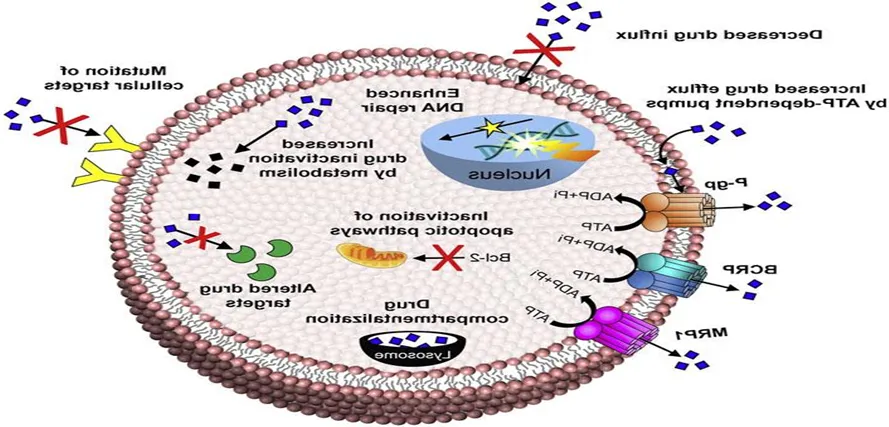
Figure 7. Presence of Efflux Pumps
A drug must remain inside a bacterial cell at high levels for an extended time to have a lasting effect. However, certain bacteria use highly effective drug efflux pumps to remove the drug quickly once it enters the cell, leaving only minimal traces that are not enough to cause any meaningful effect. Some of these pumps specifically target certain types of antibiotics, such as macrolides, lincosamides, streptogramins, and tetracyclines, while others can remove a wide range of drugs with different structures and functions. Most drug efflux proteins belong to five main families: resistance-nodulation-cell division (RND), major facilitator (MF), staphylococcal/small multidrug resistance (SMR), ATP-binding cassette (ABC), and multidrug and toxic compound extrusion (MATE). Except for ABC transporters, the efflux mechanisms of the other families are powered by proton or sodium motive forces, and this is known as secondary transport. In contrast, primary ABC transporters use ATP hydrolysis to drive drug efflux. These mechanisms have been seen in:
(a) E. coli and other Enterobacteriaceae against tetracyclines
(b) Enterobacteriaceae against chloramphenicol
(c) Staphylococci against macrolides and streptogramins
(d) Staphylococcus aureus and Streptococcus pneumonia against fluoroquinolones
Key features
Decreases intracellular drug accumulation
Causes resistance to multiple drugs (multidrug resistance)
Affects drugs like Doxorubicin and paclitaxel[10].
4.2 DNA Repair Mechanisms
DNA repair mechanisms are essential in drug resistance during cancer therapy, as chemotherapeutic drugs such as cisplatin cause DNA damage to trigger apoptosis in cancer cells. Resistance may develop when cells have operational DNA repair mechanisms, such as nucleotide excision repair (NER) and homologous recombination repair (HR), that oppose the drug's impact. Disabling these repair mechanisms increases drug sensitivity. Mutations and epigenetic changes might make these pathways dysfunctional, making them possible therapeutic targets. Increased DNA repair processes, particularly those involving alkyltransferase, can provide resistance to drugs like doxorubicin. Normal cells control the cell cycle to enable DNA repair during the G1/S, intra-S, and G2/M checkpoints, whereas cancer cells frequently evade these regulations, resulting in modified repair processes. Cell cycle arrest facilitates damage repair, making it optimal to pair DNA-damaging agents with repair inhibitors. Impaired DNA repair systems in cancer may result in reliance on alternative methods, which are less prevalent in healthy cells and can be exploited. Crucial components, like ERCC1 in the NER pathway, are frequently overexpressed in particular cancers, enhancing resistance to chemotherapy. Research indicates that signaling pathways, including ERK1/2 and p38, play regulatory roles in the expression of ERCC1. Mismatch repair systems preserve genomic stability, and their deficiencies can lead to resistance against platinum drugs. The increased expression of miRNA-21 might hinder MMR proteins, affecting the efficiency of DNA damage repair [10, 11].
4.3 Inhibition Of The Cell Death
Cell death is mediated by three key processes: necrosis, apoptosis, and autophagy. These processes differ in their biological characteristics, yet all contribute to cell death. Apoptosis can occur through both internal and external pathways. In the external pathway, ligands and death receptors, such as FAS and TNF-R, along with linker proteins, caspases-3, -6, -7, and -8, are involved. This leads to the proteolysis of actin and nuclear lamin proteins, which ultimately results in apoptosis. In the internal pathway, which occurs in the mitochondria, proteins such as Bcl2 and AKT act as anti-apoptotic factors, while Bax, Bak, and caspase-9 function as pro-apoptotic proteins. The up-regulation of anti-apoptotic genes like Bcl2 and AKT, and the down-regulation of pro-apoptotic genes like Bax and Bclxl, are linked to increased resistance to chemotherapy. Additionally, mutations in the p53 gene can lead to drug resistance, as these mutations impair the connection between DNA damage caused by chemotherapeutic agents and the activation of apoptosis[12, 13].
4.4 Epigenetic Altering Cause Drug Resistance
These alterations include DNA methylation and histone modifications. DNA methylation involves the addition of a methyl group to the 5' carbon of cytosine in CpG islands, typically located upstream of gene promoters. However, methylation can also occur in other genomic regions. Histone acetylation and deacetylation, mediated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively, alter the structure of chromatin. Acetylation of lysine residues leads to an open chromatin structure, while deacetylation causes chromatin condensation, thereby affecting gene expression. For instance, tumor suppressor genes are often silenced by methylation, while hypermethylation of oncogenes can enhance their expression. Demethylation of the MDR1 gene in cancer cells leads to the development of multi-drug resistance, reducing the intracellular accumulation of anti-tumor drugs. MDR1 is overexpressed in immature myeloid cancer cells but is downregulated in mature cells.
Epigenetic changes can also influence DNA repair mechanisms. The mismatch repair system involves proteins such as hMLH1 and hMSH1, and mutations or hypermethylation in the promoter region of these genes can contribute to cancer development. For example, mutations or hypermethylation of the hMLH1 gene can lead to colorectal cancer. Decitabine (DAC), an inhibitor of DNA methylation, does not directly affect tumor growth but can increase sensitivity to other drugs, such as cisplatin and carboplatin. Similarly, demethylation of the hMLH1 promoter by DAC can restore the mismatch repair system, making colorectal cancer cells more responsive to 5-FU. Combining epigenetic agents with traditional chemotherapeutic drugs can be effective in treating drug-resistant tumors and cancer cells.[14, 15].
5 Key Signaling Pathways in Drug Resistance
5.1 PTEN/PI3K/Akt pathway
This pathway is involved in controlling many important physiological and pathological processes such as cell proliferation, angiogenesis, metabolism, differentiation, and cell survival. Mutations in the phosphatase and tensin homolog (PTEN) gene are commonly observed in more than half of the glioblastoma multiforme specimens. Interestingly, stemness genes such as OCT4, SOX2, and NANOG are expressed in PTEN-knockout neural stem cells. These cells still have the ability to differentiate into various cell lineages, but they also develop a neoplastic phenotype, including increased growth, resistance to cell death, and enhanced migratory potential, along with in vivo invasiveness.[16]. One of the key roles of the PI3K/Akt pathway is controlling the activity of ABCG2 by positioning it at the cell surface.
The side population characteristics of glioma cancer stem-like cells are enhanced as a result of PTEN inactivation. Regarding the role of the Notch pathway in the resistance of glioma stem cells, inhibitors of γ-secretase increase the susceptibility of these cells to treatment. This is due to the activation of the PI3K/Akt pathway and the overexpression of a truncated form of the apoptotic protein Mcl-1. Stromal-derived factor 1a and its receptor CXCR4 are involved in the migration of blood-forming cells to the bone marrow. The interaction between this ligand and its receptor plays a significant role in the resistance of leukemia cells to apoptosis following chemotherapy. As a result, AMD3100, which inhibits CXCR4, prevents the phosphorylation of Akt and stops the cleavage of PARP, thus increasing the sensitivity of cancer cells to chemotherapy in leukemia. Since the mobilization of hematopoietic stem cells depends on CXCR4, blocking this receptor makes multiple myeloma cells more vulnerable to chemotherapy by inhibiting their adhesion to stromal cells in the bone marrow. In a manner similar to the PI3K/Akt pathway, PD-L1 influences stemness markers such as OCT-4, NANOG, and BMI1 in breast cancer.[17, 18].
5.2 NF kB pathway
NF-kB plays a role in regulating both innate and adaptive immunity. It is essential for inflammatory responses. NF-kB is involved in the survival, activation, and differentiation of cells. Activation of NF-kB increases the production of interleukin 3 (IL-3) and granulocyte-macrophage colony-stimulating factor (GM-CSF), which in turn promote the proliferation and survival of stem cells in leukemia. In breast and lung cancer, the NF-kB signaling pathway promotes the interaction between LIN28 and TCF4, which supports stemness and metastasis. Breast cancer cells and cancer stem cells (CSCs) express high levels of IL-8 after chemotherapy, which forms an inflammatory loop between NF-kB and the STAT3 signaling pathways, as shown by Rezayatmand et al. in Stem Cell Research & Therapy (2022) 13:181, Page 5 of 16. Blocking Toll-like receptor-7 reduces the growth rate of CSCs in hepatocellular carcinoma by targeting the TLR7- IKK-NF-kB-IL6 signaling pathway [19].
5.3 P53 Pathway
p53 is a transcription factor and a tumor suppressor that becomes active under cellular stresses such as DNA damage, oxidative stress, and lack of nutrients.
p53 is activated through both phosphorylation and acetylation, which disrupt its interaction with negative regulators, increase its stability and DNA binding ability, and enable it to bind transcriptional co-activators to regulate gene expression. The range of genes regulated by p53 is continuously expanding; the most well-known functions of p53 are in promoting cell cycle arrest, apoptosis, or cellular senescence in damaged cells. The name p53 was derived from the observed molecular weight (53 kilodaltons) when the protein was run on an SDS-PAGE gel.
Cancer Impact :- p53 is the most commonly mutated gene in human cancers, with more than 50% of these mutations being missense mutations in the DNA-binding domain, often at one of six specific sites.These mutations can be inherited or occur due to exposure to mutagens such as radiation or viruses, including human papillomavirus (HPV). These mutations impair p53's ability to activate gene transcription and also exert a dominant negative effect on the function of normal p53 through oligomerization. Specifically, the loss of p53's pro-apoptotic function is crucial for the development of cancer. Inheriting only one copy of the functional p53 gene significantly reduces its tumor-suppressive activity, leading to the development of tumors during early adulthood, a condition known as Li-Fraumeni syndrome. Recent studies have also identified gain-of-function effects associated with p53 mutations, expanding the known mechanisms by which mutant p53 contributes to cancer progression.
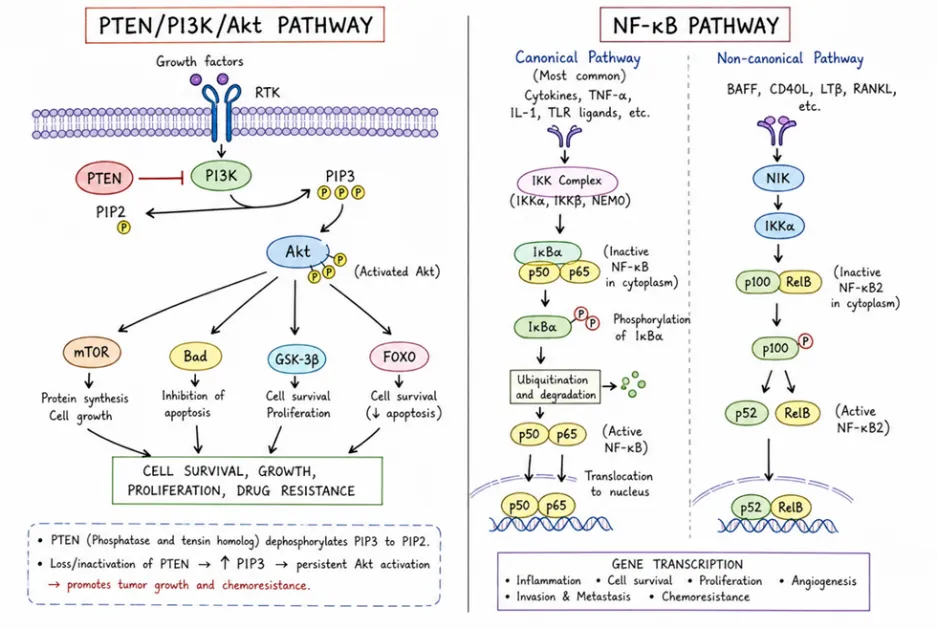
Figure 8. PTEN/PI3K/Akt Pathway & NF Kb Pathway
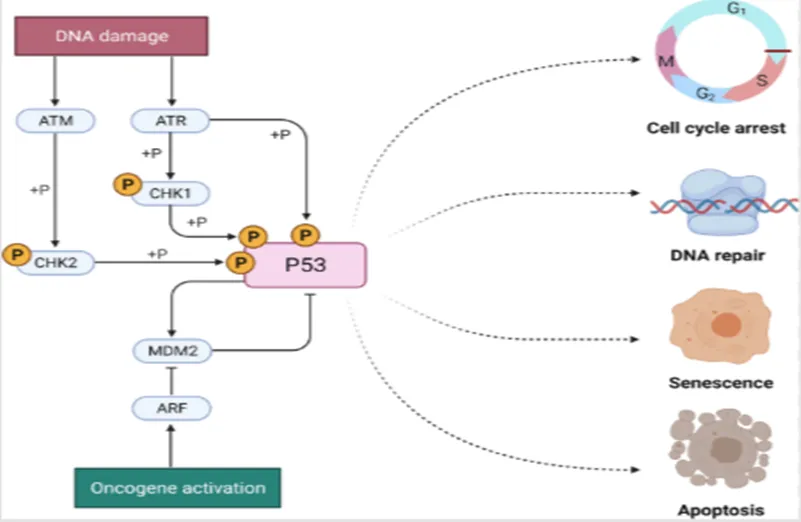
Figure 9. p53 signaling in cancer progression and therapy
6 Tumor Microenvironment (TME) & Cancer Stem Cells & their Drug Resistance
6.1 Tumor Microenvironment (TME)
6.1.1 Hypoxia :-
Oxygen deprivation, or hypoxia, affects cells and tissues, with solid tumors often existing in hypoxic states. Cancer cells respond to hypoxia by either slowing growth, leading to cell death, or by adapting. Hypoxia-inducible factors (HIFs) are crucial proteins that facilitate survival under low oxygen. HIFs contain an α subunit, typically inactivated by prolyl hydroxylase dioxygenase (PHD) in normoxic conditions. In hypoxia, the α subunit binds to the β subunit, allowing the complex to enter the nucleus; there are three HIF-alpha types and one HIF-beta type. HIF-1 alpha is particularly important for cancer cell resistance to chemotherapy. Standard cancer treatments utilize pathways that induce cell death, yet the tumor microenvironment, especially in hypoxic conditions, can foster treatment resistance and promote cancer growth and spread. Treatments like gemcitabine and doxorubicin require oxygen for effectiveness, which diminishes in hypoxia. Furthermore, hypoxic cells experience limited drug access due to poor vascularity and display reduced drug bioavailability and altered cellular mechanisms. Pre-incubation in hypoxic conditions significantly amplifies resistance to various drugs, demonstrated in both in vitro and in vivo models.[20-22].
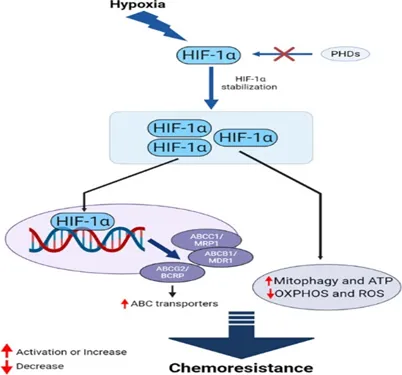
Figure 10. HIF-1α mediates the chemoresistance features of cancer cells through multiple and interconnected mechanisms
6.1.2 Stomatal cells
Cancer cells can recruit cells from the surrounding tissue to help them grow and form tumors. The types of these cells can vary depending on the type of tumor and include blood vessel cells, fibroblasts, fat cells and stellate cells. Once these cells are present in the tumor environment they release factors that influence the formation of blood vessels, cell growth, invasion and spread.[23].
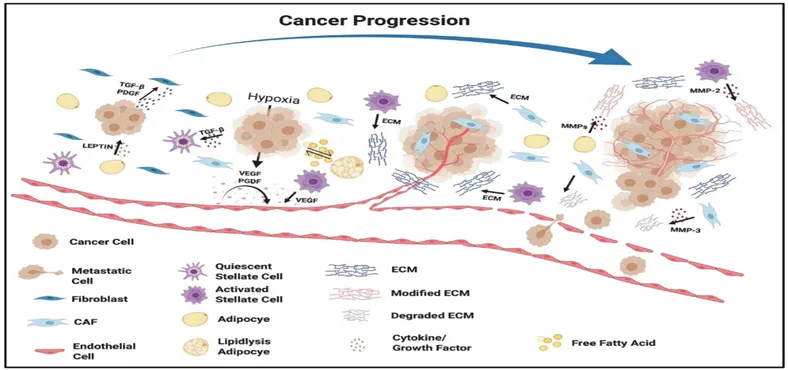
Figure 11. Defining the role of stromal cells in promoting cancer progression.
6.1.3 Angiogenesis
Angiogenesis refers to the formation of new blood vessels, primarily through the sprouting of existing vessels. Endothelial cells (ECs) that line the inner surface of blood vessels are usually in a quiescent state but can be activated during development, wound healing, or disease to enter the angiogenic process. Tumors start in a pre-vascular phase, where the diameter is less than 1 mm and there are no vessels, with cells relying on diffusion to obtain nutrients. As the tumor grows, new vessels develop through angiogenesis, allowing the tumor to acquire nutrients. If angiogenesis does not occur, the lack of oxygen and other nutrients limits the tumor size to 2-3 mm.[24]. Studies by Li and colleagues showed that hypoxia is closely related to blood flow. In a model of peritoneal metastatic colorectal cancer, they found that small microscopic tumors were not connected to blood vessels and were very hypoxic, while larger tumors (1-4mm in size) had developed vessels and were less hypoxic. As tumors grow, their rapid expansion outpaces the local blood supply, and the inefficient vascular network cannot provide enough oxygen and nutrients, resulting in hypoxia in larger tumors.
Angiogenesis is activated in response to hypoxia through the HIF1 pathway, leading to the production of VEGF by both cancer and surrounding cells. Vascular endothelial cells in the tumor have distinct genetic profiles and surface receptors, resulting in abnormal vessel structures. This irregular vascular network is characterized by leaky and compressed blood and lymphatic vessels, which lead to further regional hypoxia, creating a cycle that worsens the situation. As a result, cancer cells experience cycles of hypoxia and reoxygenation, which may cause more aggressive cancer behavior.[25]
6.1.4 Immune suppression
Cancer-associated inflammation plays a role in all stages of tumor growth. It helps cause changes, cell growth and new blood vessel formation. This inflammation also helps cancer cells avoid death and spread to parts of the body. When oxygen levels are low cancer cells become resistant to the system. They can evade detection by the system. Low oxygen levels are controlled by factors called Hypoxia-inducible factors (HIFs)[26]. These factors affect how genes related to the system are expressed. Areas of tumors with oxygen have a lot of certain types of immune cells. These include derived suppressor cells (MDSCs) tumor-associated macrophages (TAMs) and regulatory T cells (Tregs). Low oxygen for a time makes it harder for certain immune cells to kill cancer cells. These immune cells include killer (NK) cells and CD8 T cells. High levels of lactate in tumors also stop these cells from working properly[27]. This makes it harder for the system to fight cancer. Research shows that reducing oxygen levels can help restore the function of neutrophils to fight tumors. Low oxygen levels increase the production of PD-L1 in cancer cells and immune cells[28]. This blocks responses. Low oxygen also affects B cells causing them to divide less die more and make antibodies. This impacts the systems ability to remember and fight cancer.The effects of oxygen, on dendritic cells are not fully understood.. Some evidence suggests that HIF-1α may help dendritic cells trigger immune responses. It may affect their survival, movement and ability to present antigens[26, 29].
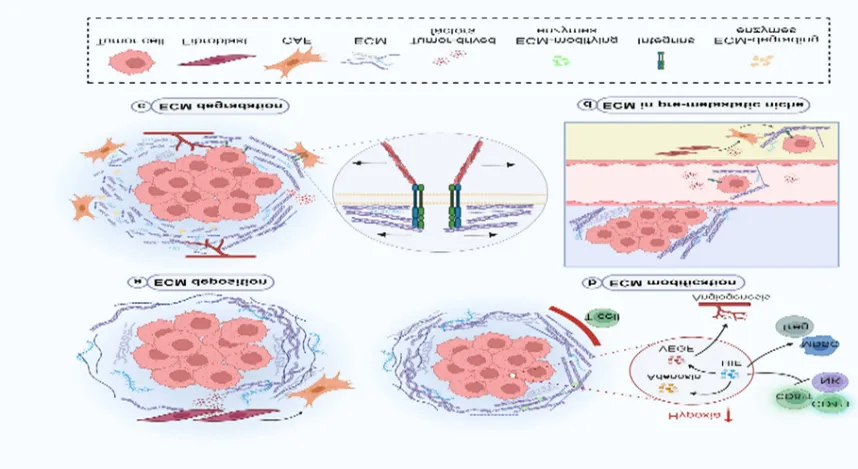
Figure 12. From: Immunosuppressive tumor microenvironment in the progression, metastasis, and therapy of hepatocellular carcinoma: from bench to bedside
6.2 Cancer Stem Cells
cancer stem cells (CSCs), also known as tumor-initiating cells (TICs), have been intensively studied in the past decade, focusing on the possible source, origin, cellular markers, mechanism study, and development of therapeutic strategy targeting their pathway .The first convincing evidence of CSCs was reported by Bonnet and Dick in 1997 by the identification of a subpopulation of leukemia cells expressing surface marker CD34, but not CD38. CD34+/CD38− subpopulation was capable of initiating tumor growth in the NOD/SCID recipient mice after transplantation .In addition to blood cancer, CSCs have been identified in several kinds of solid tumor . The first evidence of the presence of CSCs in solid cancer in vivo was found and identified as CD44+CD24-/lowLineage− cells in immunocompromised mice after transplanting human breast cancer cells in 2003 even though it has been indicated in vitro in 2002 by the discovery of clonogenic (sphere-forming) cells isolated from human brain gliomas . Over time, CSC population was also identified from several other solid cancers including melanoma, brain, lung, liver, pancreas, colon, breast cancer, as well as ovarian cancer [30, 31]. Although CSC model explains the heterogeneity of cancers in terms of hierarchical structure and progression mode, the origins of CSCs are currently unclear and controversial .Accumulating hypotheses suggest that depending on the tumor type, CSCs might be derived from either adult stem cells, adult progenitor cells that have undergone mutation, or from differentiated cells/cancer cells that obtained stem-like properties through dedifferentiation .Because of the plasticity of CSCs, it has been suggested that the combinational therapy of targeting CSC pathways and conventional chemotherapeutics might have better therapeutic effect, which will be explained later in detail (Figure 1). Early studies in AML demonstrated that normal primitive cells, but not committed progenitor cells, are targets for leukemic transformation [30]. Similarly, it has been indicated that deletion of Apc in Lgr5+ (leucine-rich-repeat containing G-protein coupled receptor 5) long-lived intestinal stem cells, rather than short-lived transit-amplifying cells, could lead to their transformation, showing that stem cells are the cells-of-origin in intestinal cancer [32]. Moreover, long-term culture can also induce telomerase-transduced adult human mesenchymal stem cells (hMSCs) to undergo spontaneous transformation, showing that these cells are also the origin of CSCs . Interestingly, CSCs originate from the transformation of not only their tissue-specific stem cells but also other tissue stem cells. For instance, bone marrow-derived cells (BMDCs) may be an essential source of many tumor types, such as gastric cancer, neural tumors, and even teratoma [33].

Figure 13. The origin of CSCs and combinational therapy of CSC targeting and bulk tumor ablation.
Role in Cancer Relapse
Cancer Stem Cells have an ability to renew and change into different types of cells. This helps them to regrow the tumor after treatment has killed most of the non-stem cancer cells. Many Cancer Stem Cells stay dormant. Do not divide. Since most treatments target dividing cells these dormant cells survive and come back later to cause the cancer to relapse. Cancer Stem Cells can also help cancer spread. They can change into a form that allows them to break away from the tumor travel to other parts of the body and form new tumors that are hard to treat.
Drug Resistance Properties
Cancer Stem Cells have pumps that can push chemotherapy drugs out of the cell.They are also good at fixing damage to their DNA. This helps them to survive when they are damaged by radiation or chemotherapy.Cancer Stem Cells have signals that prevent them from dying.The area, around the Cancer Stem Cells also helps to protect them. It acts like a shield that keeps them safe from treatments.Cancer Stem Cells have proteins that help them to stay alive. They use pathways in the body that help them to survive. These pathways include Wnt and beta catenin, Notch, Hedgehog and PI3K/Akt.
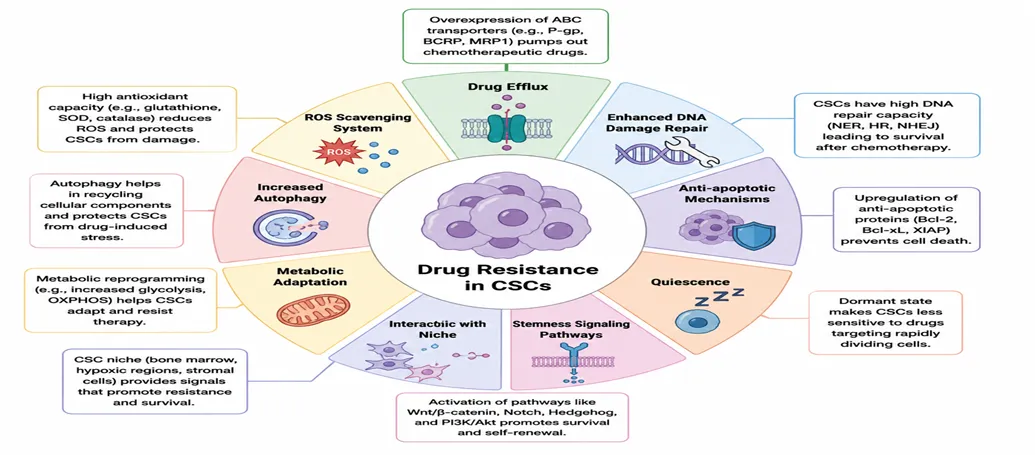
Figure 14. Schematic presentation of CSC-mediated therapy resistance to cancer.
7 Pharmacological Mechanisms of Anticancer Drugs with Clinical Insights: Focus on Cisplatin Therapy in Ovarian Cancer
7.1 Common anticancer drugs and their mode of action
Anticancer drugs play a crucial role in the treatment of various malignancies. Chemotherapy, which targets fast-growing cells like cancer cells but also harms healthy cells and causes severe side effects, is a treatment option for cancer. Several chemotherapeutics are frequently combined to address issues like drug resistance, molecular instability, and poor water solubility, which limits cell membrane permeability. Other strategies involve small molecules like genes, small RNAs, and plasmids, though these also face challenges due to poor stability in vivo [13]. The most significant clinical advances in anticancer therapeutics have emerged from treatments with novel mechanisms of action. New classes of cytotoxic chemotherapies, such as anthracyclines and taxanes, have notably improved outcomes across various tumor types. Imatinib, a pioneering kinase inhibitor, has revolutionized the treatment of chronic myeloid leukemia. Additionally, monoclonal antibodies have made substantial progress by effectively targeting CD20 in B-lymphoma, ERBB2 in breast cancer, and immune checkpoints, leading to enhanced therapeutic efficacy [14,15,16]. Here, a number of anticancer drugs are listed according to their class (Table 1). Their mode of action and the types of cancer that are used to treat are also mentioned below. Table 1 List of anticancer drugs and their mode of action Class Drug Name Mode of Action Cyclophosphamide Cross-links DNA, leading to DNA strand breaks and apoptosis. Cancer Types Treated Lymphomas, leukemias, breast cancer Cisplatin Forms DNA adducts, causing DNA cross-linking and strand breaks. Ifosfamide Testicular, ovarian, bladder cancer Similar to cyclophosphamide; forms DNA cross-links, leading to cell death. Alkylating Drugs Melphalan Sarcomas, lymphoma and lung cancer. Alkylates DNA, leading to crosslinking and apoptosis. Chlorambucil Multiple myeloma, ovarian cancer Cross-links DNA, interfering with DNA replication and transcription. Busulfan Chronic lymphocytic leukemia, lymphomas Cross-links DNA, leading to DNA damage and cell death. Carmustine (BCNU) Alkylates DNA and RNA, causing cross-linking and strand breaks. Chronic myeloid leukemia and myelofibrosis Brain myeloma tumor, multiple
Table 1. Anticancer Drugs — Complete List with Mode of Action
|
Drug Class |
Drug Examples |
Primary Mode of Action |
|
Alkylating Agents |
Cyclophosphamide, Melphalan, Cisplatin, Carboplatin |
Damages DNA by attaching alkyl groups, preventing cell replication |
|
Antimetabolites |
Methotrexate, 5-Fluorouracil, Gemcitabine, Capecitabine |
Mimics building blocks of DNA/RNA, interrupting synthesis |
|
Plant Alkaloids |
Paclitaxel, Docetaxel, Vincristine, Vinblastine |
Interferes with microtubule formation/function during mitosis |
|
Antitumor Antibiotics |
Doxorubicin, Bleomycin, Mitomycin |
Intercalates with DNA, inhibiting DNA/RNA synthesis |
|
Topoisomerase Inhibitors |
Etoposide, Irinotecan, Topotecan |
Inhibits topoisomerase I/II enzymes, causing DNA strand breaks |
|
Targeted Agents |
Bortezomib, Olaparib, Trastuzumab |
Inhibits specific proteins/pathways necessary for tumor growth |
7.2 Case Study :- Cisplatin Therapy In Ovarian Cancer
7.2.1 Introduction
Cisplatin is a chemotherapeutic drug that has been used in the treatment of various types of human cancers such as ovarian, lung, head and neck, testicular and bladder. Cisplatin is a chemotherapy medication used to treat patients with bladder, ovarian, head and neck, lung, testicular, cervical, esophageal, breast and brain cancers often given as an injection. Cisplatin, referred to by chemical name as cis-diamminedichloroplatinum (II), is anticancer, neutral and DNA destroying agent that is square planar platinum (II) complex and contains 2 ligands of chloride in a cis configuration orientation.Cisplatin has been cited as being among the most used cytotoxic anticancer medication due to its broader efficacy in the treatment of various types of cancers.The mode of administration of cisplatin is intravenous as a short term infusion with normal saline for the treatment of solid malignancies. Cancer refers to the abnormal cell division with potential to invade adjacent cells. Genotypic expression of some important properties in the cell is obligatory for Carcinogenesis.Cancer also refers to the malignant neoplasm which occurs when the cellular proliferation in the normal body tissues is no longer under normal control[34]. Evidence from past studies have singled out testicular cancer as the type of cancer that cisplatin is most effective at treating. One important property of cisplatin is that it is not stable in Dimethyl Sulfoxide (DMSO) thus researchers and medical practitioners have to pay careful attention on how long it remains in the solvent. DMSO is a colorless solvent that dissolves nonpolar and polar compounds.
The administration of cisplatin among cancer patients is highly controlled due to the fact that it is associated with various side effects. High doses of cisplatin is linked to nephrotoxicity, hence practitioners are advised to reduce doses when a patient renal function is reduced. It is therefore important to note that nephrotoxicity is a dose limiting side effect.1 Evidence from nerve conduction studies done before and after cisplatin treatment also reveals that there are neurological effects such as impaired hearing and perception in some patients. Other side effects such as nausea as well as vomiting are mostly prevented by prophylactic antiemetic together with corticosteroids. The severity of the side effects differs from one patient to the other and practitioners and researchers are encouraged to be careful when administering the drug. Recent studies have suggested that combination of cisplatin therapies with other drugs are effective in not only overcoming drug resistant, but also in reducing toxicity[35].
7.2.2 Mechanism of action :- in case of Cisplatin in Ovarian Cancer
Cisplatin is administrated to patients intravenously, as a solution in saline. While circulating in the bloodstream, the drug can be easily bonded by amino acids and proteins that are present in the blood (for example, cysteine or albumin), which results in the inactivation of 65–95% of the cisplatin administered to the patient’s body. Cisplatin that has not been deactivated is imported into cells by the membrane transport proteins CTR1 and CTR2 and by passive diffusion. Once inside the cells, cisplatin is activated by exchanging one or two chloride ligands for water[36]. The hydrated and activated cisplatin is a compound with a multidirectional cytotoxic activity, including inducing oxidative stress in cells, as well as damaging cellular and mitochondrial DNA in cancer cells [37].
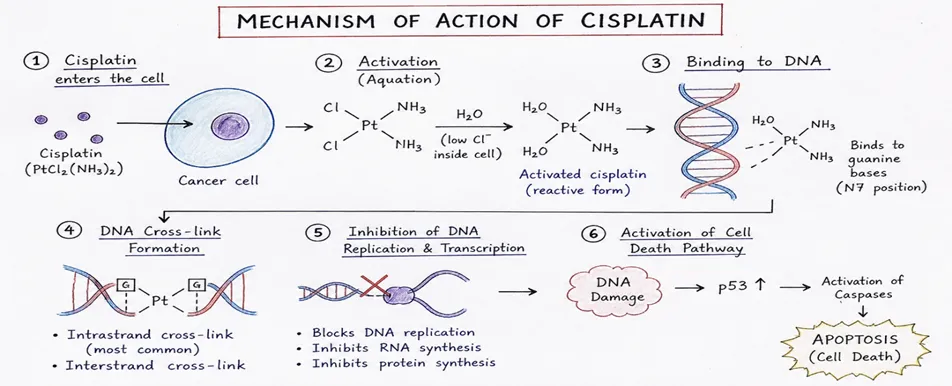
Figure 15. Mechanism Of Action
7.2.2.1 Damaging the Genomic DNA
Cisplatin binds preferentially to N7 positions of guanine and adenine, forming: intrastrand adducts (65% GG, 25% GA on same strand) and interstrand crosslinks (5%). DNA damage activates NER and MMR repair systems; when repair fails, apoptosis is triggered via G1/S, S, or G2/M checkpoint inhibition, halting DNA synthesis [38, 39].
7.2.2..2. Damaging the Mitochondrial DNA
Cisplatin also damages mitochondrial DNA (mtDNA), altering mitochondrial membrane permeability. This releases cytochrome C and procaspase-9, which with APAF-1 and ATP form the apoptosome, activating caspase-9 and subsequently caspases 3, 6, and 7, inducing apoptosis via the intrinsic pathway [40, 41].
7.2.2.3. Induction of Oxidative Stress in Cells
Cisplatin increases reactive oxygen species (ROS)—hydroxyl radicals and superoxides—in the cell membrane, cytoplasm, and organelles. At the cell membrane, ROS activate acid sphingomyelinase, producing ceramides that cluster FAS receptors, inducing apoptosis. In mitochondria, ROS interact with pro-apoptotic BAX, disrupting mitochondrial membrane potential and activating the intrinsic apoptosis pathway[42].
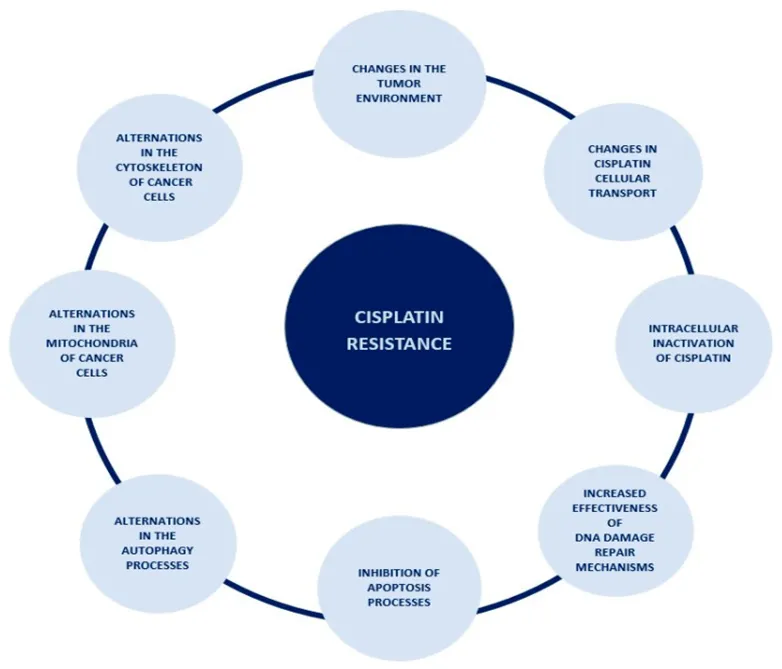
Figure 16.The spectrum of occurrences lying behind the mechanism of cisplatin resistance.
7.3 Toxicity of Cisplatin
Significant dose-dependent toxicities limit the clinical utility of cisplatin:
• Nephrotoxicity (dose-limiting): Oxidative damage is caused when cisplatin builds up in renal tubules. Patients with impaired renal function must reduce their dosage.
• Neurotoxicity includes seizures, cognitive impairments, ototoxicity (hearing loss), and peripheral neuropathy (numbness, tingling). more frequent when dosages are higher.
• Gastrointestinal toxicity: severe nausea and vomiting (average 11 episodes/dose); currently treated with corticosteroids and 5-HT3 antagonists, although approximately 50% still have nausea.
• Cardiotoxicity: Myocarditis, heart failure, and arrhythmias brought on by oxidative stress on cardiomyocytes.
• Hepatotoxicity: Hepatic oxidative stress, enzyme leakage, and inflammation are brought on by high doses of cisplatin [43].
7.4 Strategies to Increase the Effectiveness of Cisplatin in the Treatment of Ovarian Cance
Due to the growing problem of the development of resistance to cisplatin by ovarian cancer cells, methods of increasing the efficiency and effectiveness of the cytotoxic effect of cisplatin, as well as methods of inhibiting the mechanisms of resistance developed by cancer cells and restoring the sensitivity of these cells, are intensively sought. A particularly interesting strategy seems to be combination therapy—a therapy consisting of combining two or more agents with cytotoxic activity, for example, several drugs, or a drug with ionizing radiation or high temperature.The examples of potential combination therapies with cisplatin were further discussed in the following subsections and summarized in Table 1. Although platinum compounds are still the main therapy for ovarian cancers, it is absolutely important to note that histopathological heterogeneity guides the therapeutic approach in a very important way. For example, in the case of mucinous ovarian carcinoma or ovarian clear cell carcinoma, which usually demonstrates a poor response to cisplatin treatment, the molecular therapy targeting VEGF (e.g., Bevacizumab), EGFR (e.g., Gefitinib), SCR pathway (e.g., Dasatinib) or Her2 receptor (e.g., Trastuzumab) seems to be more suitable
7.4.1. Creation of Cisplatin Variants
Table 2. Cisplatin Analogus
|
Drug |
Status |
Key Indications |
Primary Toxicity |
|
Cisplatin |
Approved (global) |
Ovarian, testicular, lung, bladder, head & neck |
Nephro-, neuro-, ototoxicity |
|
Carboplatin |
Approved (global) |
Ovarian, lung, breast, bladder |
Myelosuppression |
|
Oxaliplatin |
Approved (global) |
Colorectal, gastric, esophageal |
Neurotoxicity |
|
Nedaplatin |
Approved (Japan) |
Ovarian recurrence, head & neck, esophagus, lung |
Myelosuppression |
|
Lobaplatin |
Approved (China) |
CML, breast, small-cell lung cancer |
Myelosuppression |
|
Heptaplatin |
Approved (S. Korea) |
Gastric cancer |
Nephrotoxicity |
|
Satraplatin |
Under Trial |
Prostate, non-small cell lung cancer (oral) |
Myelosuppression |
|
Picoplatin |
Under Trial |
Small-cell lung, ovarian cancer |
Myelosuppression |
7.4.2 Combination with Plant-Derived Compounds Phytochemicals represent a promising class of cisplatin sensitizers acting through independent mechanisms:
• Cardamonin (Zingiberaceae): Inhibits mTOR kinase, reverses cisplatin resistance in ovarian cancer, arrests cell cycle at G2/M.
• Berberine (Berberis vulgaris): Reduces miR-93, inactivating the AKT resistance pathway.
• Emodin (Rheum/Reynoutria spp.): Generates ROS and decreases MRP1, reducing cisplatin efflux.
• Thymoquinone (Nigella sativa): Generates ROS, inhibits NF-κB, activates tumor suppressor genes.
• Genistein (soy): Targets NF-κB pathway to enhance cisplatin sensitivity.
• Luteolin (thyme, peppermint): Induces apoptosis and inhibits angiogenesis.
• Saikosaponin-D (Bupleurum falcatum): Re-sensitizes resistant cells via G2/M arrest and intracellular calcium elevation.
7.4.3 Radiotherapy Combination
Low-dose fractionated radiotherapy enhances cisplatin sensitivity in both sensitive and resistant ovarian cancer cells. The mechanism involves upregulation of FOXO3 protein, a transcription factor governing apoptosis and metabolism, which is markedly reduced in cisplatin-resistant cells. Restoring FOXO3 levels via low-dose radiation positively influences their cisplatin responsiveness.
7.4.4 Hyperthermia Combination
Heat treatment disrupts resistance mechanisms by increasing membrane fluidity and permeability, enabling greater cisplatin cellular uptake. Hyperthermia also enhances cisplatin-DNA adduct formation, reduces glutathione levels, and inhibits DNA repair. Hyperthermic Intraperitoneal Chemotherapy (HIPEC)—administering cisplatin directly into the abdominal cavity post-surgery at 10–20× conventional concentrations—has improved survival in advanced ovarian cancer.
7.4.5 Nanocarrier-Based Delivery
Nanotechnology offers targeted drug delivery to overcome resistance mechanisms:
Lipid nanocarriers (liposomes, solid lipid nanoparticles, nanostructured lipid carriers): Deliver cisplatin directly to tumor cells; PEGylated liposomes achieve higher intracellular cisplatin levels in resistant cells.
Polymeric nanocarriers: Natural polymer-based systems enable controlled, targeted cisplatin release.
Polyamidoamine (PAMAM) dendrimers: More efficient cisplatin delivery than free drug.
Magnetic iron oxide nanoparticles: Increase intracellular ROS, reduce glutathione, and enhance cisplatin sensitivity at reduced doses.
Table 3. Examples of combination therapies with cisplatin.
|
Combination Therapy |
Possible Applications (Cancer Types) |
|
Cisplatin + Paclitaxel |
Ovarian cancer · Cervical cancer · Gastric cancer · Non-small-cell lung cancer |
|
Cisplatin + Docetaxel |
Ovarian cancer · Head and neck cancer · Non-small-cell lung cancer · Breast cancer |
|
Cisplatin + Cardamonin |
Ovarian cancer |
|
Cisplatin + Berberine |
Ovarian cancer · Breast cancer · Gastric cancer |
|
Cisplatin + Emodin |
Ovarian cancer · Non-small-cell lung cancer · Bladder cancer · Gastric cancer · Endometrial cancer |
|
Cisplatin + Thymoquinone |
Ovarian cancer · Lung cancer |
|
Cisplatin + Genistein |
Ovarian cancer · Cervical cancer · Non-small-cell lung cancer |
|
Cisplatin + Luteolin |
Ovarian cancer · Colorectal cancer |
Table 4. Summary of Approved and Under-Trial Cisplatin Analogs
|
Status |
Name |
Characteristics / Focus |
|
Approved |
Carboplatin |
Second-generation; better toxicity profile than cisplatin (less nephrotoxic); widely used in ovarian and lung cancers. |
|
Approved |
Oxaliplatin |
Third-generation; unique activity against gastrointestinal cancers; forms distinct DNA lesions that bypass cisplatin resistance. |
|
Approved |
Nedaplatin |
Second-generation; approved in Japan; similar efficacy to cisplatin but lower nephrotoxicity. |
|
Approved |
Lobaplatin |
Approved in China; used for chronic myelocytic leukemia, lung, and esophageal cancers. |
|
Approved |
Heptaplatin |
Approved in South Korea; used for gastric cancer. |
|
Under-Trial |
Satraplatin |
First oral platinum drug; Phase III trials for prostate cancer; effective in cisplatin-resistant models. |
|
Under-Trial |
Picoplatin |
Designed to overcome resistance; steric hindrance slows deactivation; in trials for small-cell lung cancer (SCLC). |
|
Under-Trial |
Lipoplatin |
Liposomal cisplatin; in Phase III trials to enhance targeting and reduce side effects. |
7.6 Conclusion :- Cisplatin Therapy in Ovarian Cancer
Cisplatin is a key anticancer drug effective against ovarian, testicular, head and neck, and bladder cancers, primarily inducing apoptosis by forming cisplatin–DNA adducts, causing oxidative stress, and damaging mitochondria. However, its use is limited by nephrotoxicity, ototoxicity, gastrointestinal toxicity, cardiotoxicity, and widespread drug resistance. Cancer cells evade cisplatin's cytotoxic effects through altered protein transporter activities that reduce drug uptake and enhance efflux, increased DNA damage repair efficiency, and modifications in cytoskeleton and mitochondria. As resistance mechanisms remain poorly understood, research is essential to enhance treatment efficacy. Efforts to increase sensitivity to cisplatin involve combination therapies with other agents like paclitaxel and docetaxel, hyperthermia, and low-dose radiotherapy. Improving drug delivery through nanocarriers offers promise, alongside cisplatin analogs like carboplatin and oxaliplatin. The article introduces innovative approaches, integrating advancements like Next-Generation Sequencing, single-cell analysis, and spatial multi-omics to enhance understanding and therapeutic strategies, especially in ovarian cancer. Continued research is vital for validating and refining these therapies.
8 Strategies to Overcome Resistance
8.1 Targeted Therapies: Combating Drug Resistance Effectively
Targeted therapies mark a significant advancement in the treatment of cancer. They provide a personalized approach by focusing on the specific molecular abnormalities and pathways that contribute to cancer progression. In contrast to traditional chemotherapy, which impacts both cancerous and healthy cells, targeted therapies are engineered to specifically inhibit or interfere with the molecules or pathways that are overactive in cancer cells. This approach is grounded in the recognition that cancer is not a single disease, but rather a collection of distinct types, each characterized by unique genetic alterations and signaling pathways. A key benefit of targeted therapies is their capacity to address drug resistance, a prevalent challenge that diminishes the efficacy of conventional treatments . By directly targeting the molecules involved in developing resistance, these therapies can either bypass or neutralize the resistance mechanisms +675 employed by cancer cells, thereby restoring the effectiveness of treatment. Several strategies assist targeted therapies in overcoming drug resistance (Figure 17).
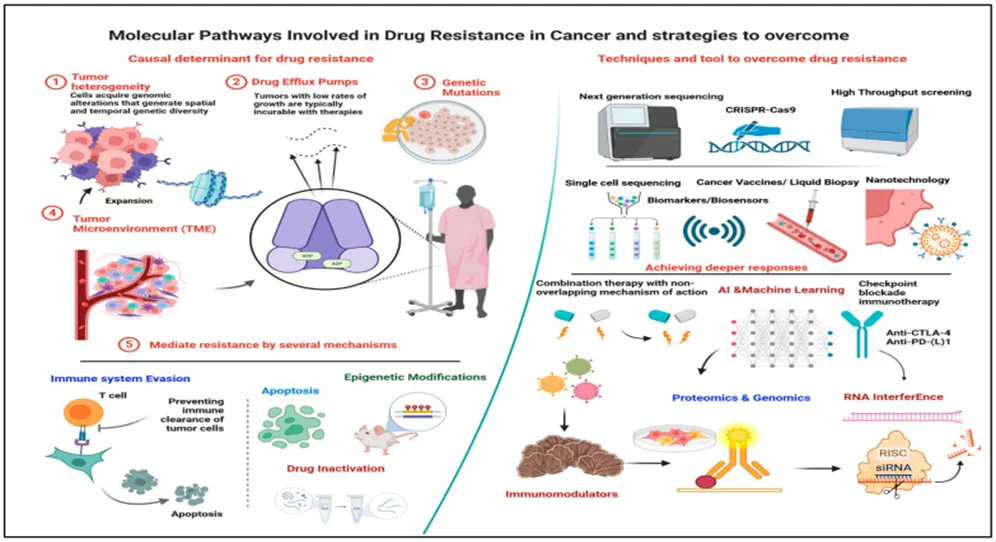
Figure 17. Molecular Pathways Of Drug Resistance In Cancer Therapy
Table 5. The commonly used methods of targeted therapy include the following:
|
Therapy Clabss |
Examples |
Mechanism/Target |
Cancer Types |
|
Tyrosine Kinase Inhibitors +TKIs) |
Imatinib, Erlotinib |
Block overactive kinases (BCR-ABL, EGFR) |
CML, NSCLC |
|
Monoclonal Antibodies |
Trastuzumab, Rituximab |
Target surface proteins; block growth signals |
HER2+ breast, B-cell lymphoma |
|
PARP Inhibitors |
Olaparib, Niraparib |
Exploit HR defects via synthetic lethality |
BRCA-mutated breast/ovarian |
|
mTOR Inhibitors |
Everolimus, Temsirolimus |
Block mTOR-driven growth/survival |
Renal, breast, neuroendocrine |
|
Proteasome Inhibitors |
Bortezomib, Carfilzomib |
Accumulate misfolded proteins → cell death |
Multiple myeloma |
|
BRAF/MEK Inhibitors |
Vemurafenib, Trametinib |
Block MAPK/ERK pathway |
Melanoma |
|
Angiogenesis Inhibitors |
Bevacizumab, Thalidomide |
Prevent tumor neovascularization |
Colorectal, ovarian, others |
|
CAR-T Cell Therapy |
Tisagenlecleucel, Axicabtagene |
Engineered T cells target tumor antigens |
Leukemias, lymphomas |
|
Checkpoint Inhibitors |
Pembrolizumab, Nivolumab |
Block PD-1/PD-L1 immune suppression |
Melanoma, NSCLC, many others |
8.2 Precision in Targeted Therapies: Overcoming Cancer Drug Resistance
Table 6. Precision in Targeted Therapies
|
Therapy Type |
What It Is |
How It Works |
Example Drugs |
Cancers Targeted |
How It Fights Resistance |
|
Tyrosine Kinase Inhibitors (TKIs) |
Drugs that block overactive enzymes (tyrosine kinases) which normally control cell growth and division — but go haywire in cancer |
Block specific tyrosine kinase proteins, cutting off the signals that tell cancer cells to grow and survive |
|
|
Directly targets the mutated or overactive kinases (e.g. EGFR or BCR-ABL mutations) that cause drug resistance in the first place |
|
PARP Inhibitors |
Drugs that block PARP enzymes — proteins the cell uses to repair damaged DNA (specifically through the base excision repair pathway) |
Use synthetic lethality: block DNA repair in cancer cells that already have a broken repair system (BRCA1/BRCA2 mutations), causing so much DNA damage the cancer cell dies |
|
|
Exploits the cancer cell's existing DNA repair defect — cancer cells with HR deficiency cannot survive when PARP is also blocked |
|
Immunotherapy (CAR-T Cell Therapy) |
A treatment that modifies the patient's own immune cells (T cells) in the lab to make them recognize and attack cancer cells |
Patient's T cells are genetically engineered to carry special receptors (CARs) that detect specific proteins on cancer cells, then destroy them |
|
|
Targets cancer antigens unaffected by conventional drugs; bypasses the immune evasion tricks used by cancer cells to hide from the immune system |
8.3 Strategic Solutions: Combating Drug Resistance with Combined Treatments.
The concept of integrating various treatment approaches to address drug resistance in cancer is grounded in the understanding that different therapies can be used together to combat resistance, enhance the effectiveness of treatment, and improve patient outcomes. This approach acknowledges that cancer cells can become resistant to a single treatment due to multiple molecular processes . Therefore, combining different treatments allows for the simultaneous targeting of multiple weaknesses within cancer cells. The reasoning behind combining treatments includes several strong justifications .
8.3.1. Targeting Diverse Pathways
Cancer cells often use multiple mechanisms to avoid the effects of therapeutic treatments.
By combining therapies that target different molecular pathways or cellular functions, it becomes more likely to overcome resistance . For example, using a tyrosine kinase inhibitor (TKI) that targets a specific signaling pathway alongside an immunotherapy agent that activates the immune system can work together to target both cancer cell growth and immune evasion .
8.3..2. Synergistic Effects
Certain drug combinations have synergistic effects, meaning their combined action is more powerful than the sum of their individual effects.
This synergy can result in stronger anti-cancer activity, increased cell death, and a reduced chance of developing drug resistance. An example is combining a DNA-damaging drug with a PARP inhibitor, which can lead to synthetic lethality in cancer cells that have defects in DNA repair, such as those with BRCA mutations .
8.3.3 Overcoming Adaptive Resistance
Cancer cells can adapt and become resistant to single treatments over time.
Using combinations of treatments with different ways of working can prevent or delay the development of resistance by targeting multiple weaknesses within the tumor. This method limits the ability of cancer cells to develop adaptive resistance mechanisms, resulting in longer-lasting treatment responses .
8.3..4. Reducing Side Effects
Using lower doses of multiple drugs that have different toxicity profiles can lower the overall side effects and toxicity compared to high doses of individual drugs.
This strategy, referred to as dose reduction or dose optimization, aims to achieve the best possible treatment effectiveness while keeping side effects manageable, thereby improving patient tolerance and quality of life during therapy .
8.4 Efficacious Therapeutic Combinations: From Clinical Trials to Preclinical Studies
The field of successful treatment combinations that have shown promise in clinical trials or preclinical studies spans a wide range of therapeutic approaches.Examining some key examples helps illustrate the potential of these combinations to transform cancer treatment.
8.4.1. Chemo-Immunotherapy Combinations
Combining chemotherapy drugs with immunotherapy, particularly immune checkpoint inhibitors such as pembrolizumab and nivolumab, has demonstrated high effectiveness in treating various cancers like melanoma, lung cancer, and bladder cancer.
These combinations utilize chemotherapy to induce a type of cell death that enhances the immune system’s ability to attack cancer cells . Clinical trials have revealed significant improvements in survival rates and treatment outcomes across different cancer types. For instance, in melanoma, using dacarbazine in conjunction with ipilimumab (an anti-CTLA-4 antibody) resulted in better results than using dacarbazine alone. Presently, research is centered on refining treatment schedules, discovering biomarkers to identify suitable patients, and testing new combinations of chemotherapy and immunotherapy to expand treatment options.
8.4.2. Targeted Therapy Combinations
The combined effect of targeted therapies with different mechanisms of action holds potential in overcoming resistance seen in cancers with specific genetic alterations.
For example, using BRAF and MEK inhibitors together in BRAF-mutant melanoma or combining EGFR and HER2 inhibitors in HER2-positive breast cancer leads to better treatment outcomes and slower development of resistance . Clinical trials have shown encouraging results in patients with these genetic changes. In HER2-positive breast cancer, adding pertuzumab (a HER2-targeted antibody) and chemotherapy along with trastuzumab significantly improved progression-free survival compared to using trastuzumab alone . Progress in therapies guided by biomarkers and precision medicine is driving the exploration of new targeted therapy combinations tailored to each patient's genetic profile.
8.4.3. PARP Inhibitor Combinations
PARP inhibitors have been effectively integrated into treatment plans alongside chemotherapy, targeted therapies, and immunotherapies for cancers with DNA repair deficiencies, such as ovarian and breast cancers with BRCA mutations.
These combinations exploit synthetic lethality, causing increased DNA damage in cancer cells and leading to improved outcomes . The use of PARP inhibitors with other treatments has been highly successful in ovarian and breast cancers. For example, combining olaparib (a PARP inhibitor) with bevacizumab (an anti-angiogenic agent) improved progression-free survival for patients with BRCA-mutated ovarian cancer . Current research is broadening the use of PARP inhibitors to other DNA repair-deficient cancers and investigating new combinations with targeted therapies and immunotherapies .
8.4.4. Dual Immunotherapy Combinations
Combining different immune checkpoint inhibitors or pairing an immune checkpoint inhibitor with other immune-modulating agents, such as cytokines or targeted therapies, shows promise in enhancing anti-tumor immune responses.
This strategy has proven effective in improving long-term survival rates in cases of melanoma and renal cell carcinoma . Clinical trials have demonstrated durable responses and improved survival rates in melanoma, lung cancer, and renal cell carcinoma with dual immunotherapy combinations. For example, combining nivolumab (anti-PD1) with ipilimumab (anti-CTLA4) in melanoma has yielded better outcomes than using either drug alone . Current research is focused on optimizing dosing regimens, identifying biomarkers to predict response, and exploring new immunotherapy combinations, including those with targeted therapies and adoptive cell therapies. The rationale behind combining different treatment modalities to combat cancer drug resistance is to target multiple pathways, achieve synergistic effects, counteract adaptive resistance, and reduce side effects. Success stories of chemo-immunotherapy, targeted therapy combinations, PARP inhibitor synergies, and dual immunotherapy underline the potential for innovative and impactful treatment strategies in cancer management[46].
8.5 Role of the Immune System in Combating Drug-Resistant Cancer Cells
Table 7. Role of the Immune System in Combating Drug-Resistant Cancer Cells
|
Mechanism / Therapy |
What it is (simple) |
How it fights drug-resistant cancer |
Key points / limitations |
|
Immune Surveillance |
The immune system constantly scans the body for abnormal cells like drug-resistant cancer cells using NK cells, dendritic cells, and macrophages. |
Finds and destroys resistant cancer cells early — before they can grow, multiply, and become harder to treat. |
Acts as a first line of defense. Works continuously in the background without any treatment being needed. |
|
Cytotoxic T Lymphocytes (CTLs) |
Specialized immune cells that directly attack and kill cancer cells by recognizing specific proteins (markers) on their surface. |
Can target cancer cells that have mutated and become resistant to drugs — recognizing new markers created by those very mutations. |
Highly precise attackers. Especially useful when cancer changes its internal pathways but still displays recognizable surface proteins. |
|
Immune Checkpoint Pathways (PD-1, CTLA-4) |
Checkpoints are like "off switches" on T cells. Cancer cells hijack these switches to hide from the immune system. |
Checkpoint inhibitor drugs block these switches, turning T cells back on so they can find and attack resistant cancer cells. |
Has transformed cancer treatment. Works even when cancer has already developed resistance to chemo or targeted drugs. |
|
Tumor-Infiltrating Lymphocytes (TILs) |
Immune cells that travel inside a tumor and work to destroy cancer cells from within the tumor environment. |
Higher numbers of TILs (especially cytotoxic T cells) inside tumors are linked to better survival and treatment outcomes in resistant cancers. |
TIL cell therapy boosts the immune system's ability to fight resistant tumors. More TILs = better prognosis in many cancers.
|
|
Cancer Vaccines |
Vaccines that train the immune system to recognize and attack specific proteins found on cancer cells — including those linked to drug resistance. |
Trigger a targeted immune response against drug-resistant cancer cells, potentially helping the body overcome resistance and improve treatment results. |
Still an emerging area. Work best when combined with other immunotherapies to strengthen the overall immune response. |
|
Immunomodulatory Therapies |
Treatments that boost or adjust the immune system's activity — making immune cells stronger and changing the tumor environment to be less protective of cancer. |
Increase immune cells' ability to kill resistant cancer, boost T cell activity, and reshape the tumor environment to support immune attacks. |
Includes CAR-T cell therapy, cytokine therapy, and targeted antibodies — often used in combination for greater effect. |
|
LAK Cell Therapy |
Patient's lymphocytes (immune cells) are collected, grown in a lab with a growth-stimulating substance (IL-2), then returned to the body as powerful "LAK cells" to attack cancer. |
LAK cells recognize and bind to tumor markers, releasing chemicals that kill cancer cells. They also stimulate other immune cells to join the attack on resistant cancer. |
Explored for melanoma, kidney cancer, and lymphoma. Limited use due to IL-2 toxicity and inconsistent results — but shows immune system potential. |
|
Targeting EMT |
EMT is a process where cancer cells change shape, become more mobile, and spread to other parts of the body — a key step in cancer progression and resistance. |
Blocking EMT pathways (TGF-β, Wnt, Notch) stops cancer cells from spreading and becoming more aggressive, reducing the chance of resistance developing. |
A targeted therapy strategy — not immune-based, but works alongside immune therapies to limit cancer's ability to adapt and resist treatment. |
8.6 Unlocking the Power of Immune Therapies: Overcoming Drug Resistance
8.6.1. Checkpoint Inhibitors
Checkpoint Inhibitors — drugs that "unmask" cancer cells so the immune system can attack them
Table 8. Checkpoint Inhibitors
|
Therapy |
How it works |
How it fights drug resistance |
Key examples / notes |
|
PD-1 / PD-L1 blockers |
Cancer cells make a protein (PD-L1) that tells immune T cells to stand down. These drugs block that signal, waking T cells back up. |
T cells become active again and can kill cancer cells even when those cells have changed to resist chemotherapy or targeted drugs. |
Pembrolizumab, Nivolumab. Work even after cancer has mutated to resist other treatments. |
|
Reversing immune suppression in tumors |
Tumors create a "shield" around themselves that exhausts nearby immune cells. |
Checkpoint inhibitors revive exhausted T cells and help them get inside the tumor, bypassing resistance linked to the tumor's protective environment. |
Especially useful when resistance is caused by suppressor immune cells (regulatory T cells, MDSCs) inside the tumor. |
|
Combination use |
Checkpoint inhibitors are paired with chemo, targeted drugs, or other immunotherapies. |
Makes cancer cells more visible to immune attacks while the inhibitor keeps T cells active — a two-pronged approach against resistance. |
Combining a checkpoint inhibitor with a drug that increases tumor antigen visibility improves patient outcomes. |
8.6.2 Adoptive Cell Therapies (ACT)
Adoptive Cell Therapies (ACT : — Patient's Own Immune Cells Are Collected, Upgraded In A Lab, Then Returned To Fight Cancer
Table 9. Adoptive Cell Therapies.
|
No |
Strategy |
How it works (simple) |
How it fights drug resistance |
Key examples / notes |
|
1 |
CAR-T cell therapy |
T cells are taken from the patient, given a new receptor (CAR) in a lab so they can recognize cancer cells, then returned to the body. |
Targets proteins on cancer cells that are unaffected by standard drug resistance — completely bypasses the usual escape routes cancer uses. |
CAR-T targeting CD19 works in patients who failed chemo. Highly effective in blood cancers like leukemia and lymphoma. |
|
2 |
Enhanced killing ability |
Engineered T cells or NK (natural killer) cells are made more powerful and precise than natural ones before being put back into the body. |
Overcomes resistance caused by cancer cells "pumping out" drugs or becoming less sensitive — the immune attack doesn't rely on a drug entering the cell at all. |
NK cells can also be used alongside T cells for broader coverage and stronger attack power. |
|
3 |
Long-lasting immune memory |
After treatment, CAR-T cells stay in the body long-term and "remember" the cancer they fought. |
Protects against cancer coming back after initial treatment — addresses resistance that develops when tumors regrow after a first response. |
If cancer returns, the immune memory allows a faster, stronger second attack compared to starting treatment from scratch. |
|
4 |
Combination with other therapies |
ACT is paired with checkpoint inhibitors or cytokine treatments to boost the immune response from multiple angles. |
Attacking multiple parts of the immune system at once makes it much harder for cancer to develop resistance to all of them simultaneously. |
Combined ACT + checkpoint inhibitor strategies outperform either therapy used alone in drug-resistant cancers. |
9 Emerging Trends
9.1 CRISPR Gene Editing
9.1.1 Introduction
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) gene-editing technology has transformed genetic engineering by providing a more efficient, precise, and scalable method for modifying genetic sequences. Initially identified as part of the bacterial immune system, CRISPR-Cas systems have now been adapted for use in eukaryotic cells, making them powerful tools for genome manipulation. This breakthrough has significantly advanced our ability to study gene function, correct disease-related genetic mutations, and develop personalized therapeutic strategies. Compared to earlier genome-editing techniques such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), CRISPR-Cas systems are more versatile and easier to use, making them a preferred approach for targeted gene modification .The CRISPR-Cas9 system, first described in 2012, is the most widely used gene-editing tool due to its capacity to introduce double-stranded breaks (DSBs) at specific genetic locations guided by a programmable single-guide RNA (sgRNA). Once the DNA is cleaved, the cell's natural repair mechanisms either repair the break using non-homologous end joining (NHEJ), which often results in insertions or deletions (indels) that can knock out a gene, or through homology-directed repair (HDR), which allows for precise gene corrections when a repair template is available. These mechanisms make CRISPR-Cas9 highly valuable in applications like gene therapy, functional genomics, and drug development.In addition to CRISPR-Cas9, other CRISPR-associated nucleases have been developed to broaden the scope of genome editing, each offering unique benefits that support precision medicine applications.
CRISPR-Cas12 and CRISPR-Cas13 are two such systems that provide distinct advantages over Cas9, particularly in terms of specificity and their ability to target RNA (Table 1) .
Table 10 . Comparison Of The Crispr-Cas9, Crispr-Cas12, And Crispr-Cas13 Systems.
|
Feature |
CRISPR-Cas9 |
CRISPR-Cas12 |
CRISPR-Cas13 |
|
Target Type |
Double-stranded DNA |
Single-stranded and double-stranded DNA |
Single-stranded RNA |
|
Cleavage Mechanism |
Creates double-strand breaks (DSBs) |
Creates single-strand staggered cuts |
Cleave RNA molecules |
|
Guide RNA (gRNA) |
Single guide RNA (sgRNA) |
Single guide RNA (sgRNA) |
CRISPR RNA (crRNA) |
|
Recognition Motif (PAM or PFS) |
PAM sequence (5′-NGG-3′) |
PAM sequence (5′-TTTV-3′) |
PFS (Protospacer Flanking Site) instead of PAM |
|
Editing Precision |
High, but off-target effects possible |
Higher specificity than Cas9 |
High specificity for RNA editing |
|
Primary Applications |
Gene knockout, gene correction, genome-wide screening |
Genome editing, diagnostics (e.g., SHERLOCK) |
RNA interference, viral RNA targeting, transcriptome regulation |
|
Advantages |
Efficient for genome editing, widely studied, well-characterized |
Higher specificity, useful for diagnostics and collateral activity, enables detection applications |
Can target RNA without affecting genome, potential for antiviral therapies |
|
Limitations |
Potential for off-target mutations requires PAM sequence |
Less well-characterized than Cas9, requires PAM sequence |
Collateral RNA degradation limits specificity and is not useful for DNA editing |
9.1.2 Mechanistic Insights into CRISPR Genome Editing :-
The CRISPR-Cas9 system has become a groundbreaking tool in genome editing due to its ability to introduce precise genetic modifications at specific genomic locations.
Unlike earlier methods that required complex protein engineering, CRISPR operates through a programmable guide RNA (gRNA) that directs the Cas9 nuclease to a complementary DNA sequence. The flexibility to target virtually any gene by adjusting the gRNA sequence has made CRISPR an essential tool in functional genomics, precision medicine, and gene therapy.
At the molecular level, CRISPR-mediated genome editing typically involves two distinct DNA repair pathways following the introduction of a double-stranded break by Cas9: Non-Homologous End Joining (NHEJ) and Homology-Directed Repair (HDR) (Figure 18).
Each pathway has different functional implications, determining whether CRISPR will be used for gene disruption, correction, or insertion of new genetic sequences. In addition to traditional CRISPR-Cas9 editing, newer advancements such as base editing and prime editing have been developed to enhance the specificity and efficiency of genetic modifications. These methods avoid the need for creating double-stranded breaks, thereby reducing off-target effects and unintentional mutations .
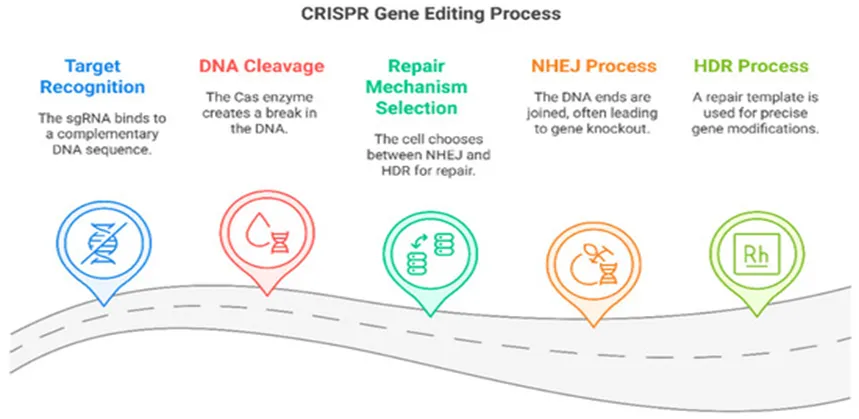
Figure 18. The CRISPR genome editing process and its mechanism of DNA modification.
1] Targeting Oncogenic Mutations for Personalized Cancer Treatment
Genetic alterations in oncogenes (KRAS, EGFR, MYC) and tumor suppressor genes (TP53, PTEN, BRCA1/2) significantly drive tumorigenesis, impacting chemotherapy resistance and cell proliferation. CRISPR enables accurate editing of these oncogenic mutations, offering prospects for personalized cancer therapies. The KRAS gene, notably mutated in pancreatic, colorectal, and lung cancers, expresses variants like KRAS G12D and G12V, which unjustly activate the RAS-RAF-MEK-ERK signaling pathway. CRISPR-Cas9 techniques, such as deletion or base editing of mutant KRAS, aim to inhibit tumor growth while preserving the wild-type allele to reduce toxicity. TP53, known as the "guardian of the genome," is frequently mutated in many cancers, disrupting DNA damage responses. CRISPR can reactivate wild-type TP53 or repair its mutations, restoring normal apoptosis in cancer cells, while synthetic lethal interactions offer insights into therapeutic vulnerabilities. EGFR mutations also drive oncogenic signaling in cancers like non-small cell lung cancer (NSCLC), with CRISPR editing employed to eliminate defective EGFR alleles and combat resistance to EGFR tyrosine kinase inhibitors. CRISPR is further being combined with immune checkpoint blockade therapies, modifying inhibitory genes in T cells (e.g., PD-1 and CTLA-4) to enhance anti-tumor immune responses, thereby improving the efficacy of immunotherapies.
2] CRISPR-Modified CAR-T Cells in Immunotherapy Chimeric antigen receptor T-cell (CAR-T) therapy represents a significant advancement in cancer immunotherapy, particularly for hematologic malignancies like acute lymphoblastic leukemia, diffuse large B-cell lymphoma, and multiple myeloma. Genetically engineered T cells, equipped with artificial receptors, target and eliminate cancer cells. Nonetheless, challenges such as immune evasion, T-cell depletion, and antigen escape complicate the therapy's effectiveness. CRISPR genome editing provides solutions to enhance CAR-T cell performance, specificity, and persistence.A significant application of CRISPR in CAR-T therapy is the deletion of immune checkpoint regulators like PD-1 and CTLA-4. Cancer cells evade immune responses by expressing PD-L1, which inhibits T-cell activity via PD-1 binding. CRISPR disruption of the PD-1 gene in CAR-T cells results in PD-1 knockout cells that show enhanced tumor-killing abilities and improved survival in the tumor microenvironment. Preclinical studies indicate that CRISPR-mediated PD-1 deletion improves responses in PD-L1-expressing cancers.
Additionally, CRISPR optimizes CAR-T cell production and safety. Traditional CAR-T therapy involves complex, patient-specific modifications, while CRISPR-Cas9 can generate off-the-shelf CAR-T cells by deleting endogenous T-cell receptor genes, reducing the risk of graft-versus-host disease and lowering production costs.
CRISPR-enhanced CAR-T therapy is also advancing solid tumor treatment, overcoming challenges due to immunosuppressive environments. Modifications improve CAR-T cell migration, survival, and ability to produce beneficial cytokines. Dual-targeting CAR-T technologies engineered through CRISPR help combat antigen escape, showing promise in treating various cancer types.
3]. CRISPR in Cancer Therapy and Precision Oncology
Cancer is a complex disease characterized by genetic and epigenetic changes that drive tumor growth, metastasis, and resistance to treatment.
Advances in genome sequencing have uncovered oncogenic mutations and dysregulated signaling pathways, allowing for the development of targeted cancer therapies. However, many cancers demonstrate genetic diversity within the tumor and can develop resistance to existing treatments over time, limiting their effectiveness. The emergence of CRISPR-Cas9 genome editing has transformed cancer research and therapy by enabling the precise modification of oncogenic mutations, tumor suppressor genes, and immune checkpoint regulators. CRISPR has become a key tool in functional
oncology, helping researchers study cancer-related mutations, identify therapeutic targets, and improve immunotherapy strategies (Figure19 )[47] .
Figure 19. AI-driven CRISPR genome editing approach for precision medicine
2 AI IN DRUG PREDICTION
1. Introduction
Artificial Intelligence (AI) refers to computer systems that can perform tasks requiring human intelligence such as learning, reasoning, and decision-making.
In drug discovery, AI is used to predict drug molecules, targets, and interactions, significantly accelerating the process. Traditional drug development takes 10–15 years and billions of dollars, whereas AI can reduce both time and cost.
2. Types of AI Used
• Machine Learning (ML) → learns from data patterns
• Deep Learning (DL) → neural networks for complex predictions
• Natural Language Processing (NLP) → analyzes scientific literature
3. Workflow of AI in Drug Prediction
Data Collection (chemical + biological data)
↓
Data Preprocessing
↓
Feature Extraction
↓
Model Training (ML/DL)
↓
Prediction (Drug–Target Interaction)
↓
Validation (In vitro / In vivo)
↓
Lead Compound Selection
4. Applications
a) Drug Target Identification
• AI identifies disease-related proteins or genes
b) Virtual Screening
• Screens millions of compounds quickly
c) Drug Repurposing
• Finds new uses for existing drugs
d) Toxicity Prediction
• Predicts side effects early
e) Molecular Design
• Generates new drug molecules
5. Advantages
• Reduces time and cost
• Handles large datasets efficiently
• High accuracy in prediction
• Improves success rate in clinical trials
• Enables personalized medicine
6. Limitations / Challenges
a) Data Dependency
• Requires large, high-quality datasets
b) Bias in Algorithms
• Biased data leads to inaccurate predictions
c) Black Box Problem
• Difficult to interpret how AI makes decisions
d) Regulatory Issues
• Lack of clear guidelines for AI-based drugs
7. Case Study / Example
• AI was used to identify potential drug candidates during COVID-19 rapidly
• Companies like Insilico Medicine use AI to design new drugs
8. Recent Advances
• Generative AI for molecule design
• AI-driven clinical trial optimization
• Integration with big data and genomics
9. Future Perspectives
• Fully automated drug discovery pipelines
• Integration with CRISPR and personalized medicine
• Faster approval of AI-designed drugs
10. Conclusion
AI in drug prediction is transforming pharmaceutical research by making drug discovery faster, cheaper, and more efficient. Despite challenges like data bias and interpretability, AI holds immense potential in the future of medicine.
9.3 PERSONALIZED MEDICINE (PRECISION MEDICINE)
1. Introduction
Personalized medicine (also called precision medicine) is an approach in which treatment is tailored to an individual patient based on their genetic, environmental, and lifestyle factors.
Unlike the traditional “one-size-fits-all” approach, personalized medicine ensures that the right drug is given to the right patient at the right dose.
2. Key Components
• Genomics → Study of patient’s DNA
• Biomarkers → Indicators of disease or drug response
• Pharmacogenomics → Effect of genes on drug response
• Companion diagnostics → Tests to guide therapy
3. Workflow of Personalized Medicine
Patient Diagnosis
↓
Sample Collection (Blood/Tissue)
↓
Genetic Testing (DNA Sequencing)
↓
Biomarker Identification
↓
Drug Selection (Targeted Therapy)
↓
Personalized Treatment Plan
↓
Monitoring & Dose Adjustment
4. Applications
a) Cancer Therapy
• Targeted therapy based on mutations (HER2, EGFR)
• Immunotherapy selection
b) Pharmacogenomics
• Dose adjustment based on metabolism
c) Chronic Diseases
• Diabetes, cardiovascular diseases
d) Rare Genetic Disorders
• Identification and treatment of inherited conditions
5. Example
• Pembrolizumab is used in patients with high PD-L1 expression
• HER2-positive breast cancer treated with targeted therapy
6. Advantages
• Increased treatment effectiveness
• Reduced adverse drug reactions
• Early disease detection
• Improved patient outcomes
• Avoids unnecessary treatments
7. Limitations / Challenges
a) High Cost
• Genetic testing and targeted drugs are expensive
b) Limited Accessibility
• Not widely available in all regions
c) Complex Data Interpretation
• Requires advanced bioinformatics tools
d) Ethical Issues
• Privacy concerns regarding genetic data
8. Recent Advances
• Next-generation sequencing (NGS)
• Liquid biopsy techniques
• Integration with AI for better prediction
9. Future Perspectives
• Routine use in clinical practice
• Integration with wearable health technologies
• Expansion in all disease areas
• More affordable genomic testing
10. Conclusion
Personalized medicine represents a major shift in healthcare by providing patient-specific treatment strategies. Although challenges like cost and accessibility remain, it has the potential to significantly improve therapeutic outcomes and revolutionize modern medicine.
10 Advantages of Understanding Resistance
Grasping the mechanisms behind cancer resistance is viewed as a significant challenge in oncology, since around 90% of chemotherapy failures and more than 50% of targeted or immunotherapy failures are linked to it. Acquiring this insight provides numerous benefits for enhancing patient results, shifting treatment from short-term relief to long-term management, or attaining cures.
Main benefits consist of:
Creation of Innovative Therapies and Combination Approaches: Comprehending resistance enables scientists to formulate new, precise medications that can evade or block particular resistance mechanisms, including increasing the specificity of antibody-drug conjugates or producing fourth-generation tyrosine kinase inhibitors (TKIs). It also aids in the creation of combination therapies—employing two or more medications—to concurrently target various pathways, hindering cancer cells from adjusting.
Enhancing Precision Medicine: By pinpointing particular genetic changes or mutations that contribute to resistance (e.g., EGFR T790M or C797S), doctors can customize therapies according to the unique molecular characteristics of the patient's tumor, steering clear of ineffective, harmful treatments.
Real-Time Surveillance and Timely Identification: Understanding the progression of cancers facilitates the creation of liquid biopsies (e.g., analyzing circulating tumor DNA or ctDNA), which can identify resistance mutations in the bloodstream prior to symptom onset, allowing for a proactive instead of a reactive strategy toward treatment recurrence.
Conquering Multidrug Resistance (MDR): A primary reason for relapse is the cancer cell's capability to expel drugs through ABC transporters. Grasping this mechanism enables the creation of inhibitors that "re-sensitize" cells to chemotherapy, ensuring existing medications remain effective for extended durations.
Focusing on the Tumor Microenvironment (TME): Resistance arises not only from the cancer cell itself but is also nurtured by its environment, including conditions like hypoxia (low oxygen) or specific cells that shield the tumor. Grasping this allows for approaches to "retrain" the TME to become less conducive to cancer persistence.
Optimizing Treatment Scheduling: Studies on resistance have revealed that some cancer cells become dependent on a drug to survive, creating vulnerabilities that can be exploited by using "evolutionary-informed" or "adaptive" therapies (e.g., intermittent scheduling) that keep the tumor manageable while reducing severe side effects.
11. Limitations
Drug resistance in cancer therapy presents several limitations that hinder effective treatment and clinical success. These challenges arise due to the complex biology of tumors and variability among patients.
11.1Tumor Heterogeneity
Tumors consist of diverse populations of cancer cells with different genetic and phenotypic characteristics
Some cells may be sensitive to drugs, while others are inherently resistant
Leads to partial treatment response and relapse
11.2 Complexity of Resistance Mechanisms
Multiple mechanisms (efflux pumps, DNA repair, apoptosis evasion, epigenetics) may occur simultaneously
Makes it difficult to target resistance with a single therapeutic strategy
Resistance pathways are often interconnected and adaptive
11.3 Development of Multidrug Resistance (MDR)
Cancer cells may become resistant to multiple structurally unrelated drugs
Commonly associated with efflux transporters and survival signaling pathways
Reduces effectiveness of combination chemotherapy
11.4 Limited Efficacy in Solid Tumors
Poor drug penetration into tumor mass
Presence of hypoxic regions and abnormal vasculature
Physical barriers reduce drug delivery to cancer cells
11.5 Toxicity and Side Effects
Increasing drug dose to overcome resistance leads to severe toxicity
Damage to normal cells limits therapeutic window
Adverse effects reduce patient compliance
11.6 Cancer Stem Cells (CSCs)
Highly resistant to chemotherapy and radiotherapy
Capable of self-renewal and tumor regeneration
Major cause of recurrence and metastasis
11.7 Lack of Predictive Biomarkers
Difficulty in predicting which patients will develop resistance
Limits personalization of therapy
Delays effective treatment decisions
11.8 High Cost and Resource Requirement
Advanced therapies (targeted therapy, immunotherapy) are expensive
Requires sophisticated diagnostic and monitoring systems
Limits accessibility, especially in developing countries
11.9 Time-Consuming Research and Development
Developing new drugs to overcome resistance takes years
High failure rates in clinical trials
Regulatory challenges further delay availability
CONCLUSION
Drug resistance in cancer treatment is a problem that affects how well current cancer therapies work. It's not one simple thing that happens but rather many factors at the molecular, cellular and environmental levels that help cancer cells survive even when they're being treated. This study shows that cancer cells are really good at adapting and use strategies to avoid being hurt by treatments. They do this by getting rid of drugs fixing damaged DNA changing what the drugs target avoiding death and changing how genes work. It's also important to know that drug resistance isn't about individual cancer cells but also about the different kinds of cells in a tumor and how they change over time. Some cells that are resistant to drugs might already be in a tumor before treatment starts. They might develop during therapy because of the pressure from anticancer drugs. This helps explain why some treatments that work at first like those using Cisplatin and Doxorubicin eventually stop working and the cancer comes back and gets worse.
The area around the tumor called the tumor microenvironment also plays a role in helping cancer cells resist drugs. This area can have oxygen levels changes in how cells use energy, a weaker immune response and interactions with surrounding tissue. Cancer stem cells make things more complicated because they can resist treatment keep themselves alive and repopulate the tumor after therapy.
From a perspective beating drug resistance means moving away from just using one drug at a time. Instead we should use combinations of drugs targeted therapies, immune-based treatments and advanced drug delivery systems that use nanotechnology. These approaches offer hope for overcoming resistance. Also getting better at diagnosing cancer at the level and finding specific markers can help design treatments that fit a patients needs at the right time.
New technologies like intelligence and CRISPR-based gene modification also offer new possibilities for predicting, tracking and managing resistance. AI can look at lots of data to predict resistance trends and gene editing tools might let us change genes involved in resistance directly. However we need to solve problems like side effects, high costs, regulatory issues and ethical concerns before these innovations can be used in medicine.
In summary drug resistance in cancer treatment is an evolving problem that requires ongoing scientific and clinical progress. Understanding the mechanisms behind resistance and using disciplines will be crucial in developing more effective long-lasting and personalized cancer treatments. Tackling drug resistance is therefore crucial, for improving survival and moving forward in the field of precision oncology.
So we can find out this project :-
• Drug resistance can be intrinsic or acquired, affecting treatment outcomes
• Multiple mechanisms act simultaneously, making resistance highly complex
• Signaling pathways such as PI3K/Akt, p53, and NF-κB play crucial roles
• Tumor microenvironment and cancer stem cells significantly contribute to resistance
• Clinical drug resistance leads to relapse, metastasis, and reduced survival
Importance in Cancer Treatment
Understanding drug resistance is essential for improving cancer therapy and patient prognosis.
• Development of targeted and personalized treatment strategies
• Use of combination therapies to overcome resistance
• Advancement in nanotechnology-based drug delivery systems
• Identification of biomarkers for predicting treatment response
• Integration of emerging technologies such as AI and gene editin
13 Future Perspectives in Overcoming Cancer Drug Resistance: Pharmaceutical Strategies and Combination Immunotherapy
Large pharmaceutical companies are implementing various strategies to address drug resistance in cancer therapy. These include the development of novel therapies to counteract the mechanisms cancer cells use to evade treatment. A pivotal approach is enhancing targeted protein degradation technologies such as proteolysis-targeting chimeras (PROTACs) and specific non-genetic IAP-dependent protein erasers (SNIPERs). These technologies diverge from traditional small-molecule inhibitors by enabling the degradation of disease-causing proteins, effectively overcoming resistance. For instance, PROTACs have successfully degraded oncogenic fusion proteins like BCR-ABL in chronic myeloid leukemia. Additionally, pharmaceutical firms are actively creating and marketing combination therapies, particularly within immunotherapy, which uses the immune system to combat tumors. By integrating immunotherapy with standard chemotherapy or targeted treatments, companies enhance treatment effectiveness and address resistance mechanisms. Through clinical trials and substantial research investments, these firms endeavor to set new standards in cancer care, reinforcing their leadership in oncology innovation and treatment progress.
• Clinical Implications and Future Directions
Grasping the mechanisms of cancer resistance and developing approaches to tackle this issue are crucial for enhancing the results of cancer treatments. Tailored treatment strategies, guided by genomic profiling and molecular diagnostics, are essential in addressing particular resistance mechanisms like drug efflux, target modifications, and DNA repair pathways. This customized method guarantees the choice of treatments that are probable to work for specific patients, thus improving treatment success rates.
In this table that shows how well these combination treatments work. It is called Table 1. We can look at this table to see what treatments work together. This helps us find the way to treat each persons cancer. Cancer treatment is very important. We need to keep working to find new and better ways to deal with it.
Table 11. Clinical outcomes of combining therapies.
|
combination Therapy |
Cancer Type |
Clinical Outcome |
|
Pembrolizumab + Chemotherapy |
Lung cancer |
Improved survival outcomes |
|
Pembrolizumab + Chemotherapy |
Bladder cancer |
Higher response rates, prolonged progression-free survival |
|
Pembrolizumab + TKIs |
Renal cell carcinoma |
Promising results in clinical trials |
|
Pembrolizumab + TKIs |
Melanoma |
Improved overall survival and response rates |
|
Trastuzumab + Pertuzumab |
HER2-positive breast cancer |
Enhanced progression-free survival |
|
Ipilimumab + Nivolumab |
Melanoma |
Increased objective response rate |
Table 12. Clinical Trials Of Combination Therapies.
|
Drug |
Combination Therapy |
Cancer Type |
Clinical Stage |
|
Pembrolizumab |
Chemotherapy (Carboplatin and Pemetrexed) |
Non-Small Cell Lung Cancer (NSCLC) |
Phase 3 |
|
Pembrolizumab |
Lenvatinib |
Renal Cell Carcinoma |
Phase 3 |
|
Trastuzumab |
Pertuzumab, Docetaxel |
HER2-Positive Breast Cancer |
Phase 3 |
|
Nivolumab |
Ipilimumab |
Melanoma |
Phase 3 |
|
Atezolizumab |
Bevacizumab, Chemotherapy (Carboplatin and Paclitaxel) |
Hepatocellular Carcinoma |
Phase 3 |
|
Durvalumab |
Tremelimumab, Chemotherapy (Cisplatin and Etoposide) |
Non-Small Cell Lung Cancer (NSCLC) |
Phase 3 |
|
Ibrutinib |
Venetoclax |
Chronic Lymphocytic Leukemia |
Phase 3 |
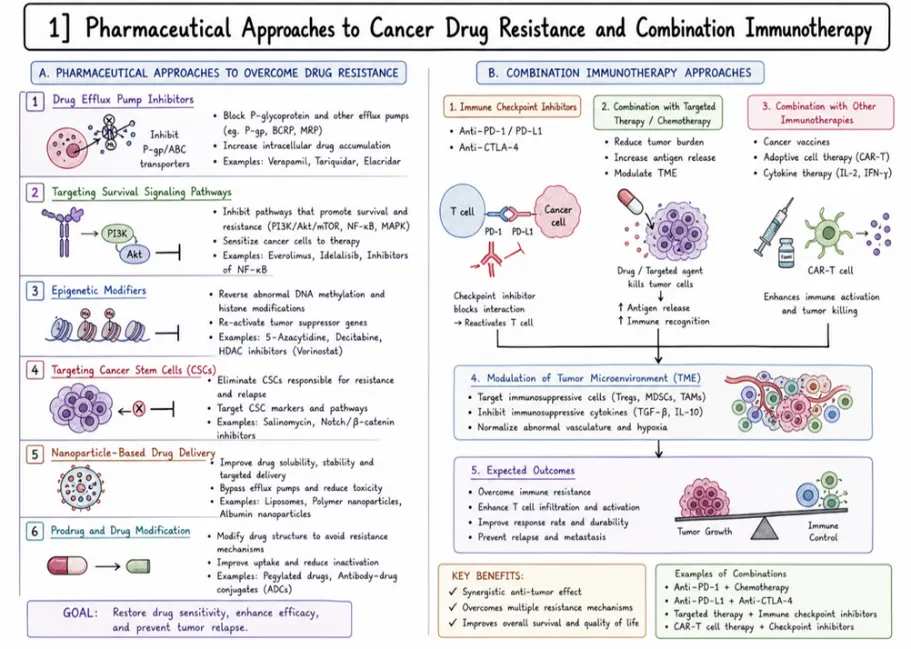
Figure 20. Pharmaceutical Approaches to Cancer Drug Resistance and Combination Immunotherapy
13.2 Future Scope: Drug Resistance in Cancer Therapy
The progress in molecular biology, biotechnology and computational sciences has opened up new ways to tackle drug resistance in cancer treatment.
Future research is focused on creating targeted and personalized methods to make treatments more effective and reduce the occurrence of resistance.
13.2.1 Personalized Medicine and Precision Oncology
Personalized medicine is a change in how cancer is treated.
The treatment is designed based on the epigenetic and molecular features of each patient.
Advances in genomic sequencing help find the mutations that lead to drug resistance.
• Using biomarkers to choose the best treatment
• Creating treatment plans to each patient
• Reducing side effects and improving treatment results
Precision oncology ensures the right drug is given to the patient at the right time.
This helps prevent the development of resistance.
13.2.2 Development of Multi-Target and Combination Therapies
Tumor cells often use signaling pathways to survive.
This makes single-drug treatment less effective.
Future strategies involve:
• Designing drugs that target pathways at the same time
• Combining treatments like chemotherapy, targeted therapy and immunotherapy in a logical way
• Stopping the tumor from finding ways to survive
These approaches greatly reduce the risk of resistance and improve treatment success.
13.2.3 Nanotechnology-Based Drug Delivery Systems
Nanotechnology provides solutions to overcome drug resistance.
It enhances drug delivery and targeting.
• The permeability and retention (EPR) effect helps drugs accumulate specifically in tumors
• Nanocarriers can bypass the mechanisms that push drugs out of cells
• slow release of the drug
• Lower toxicity to the rest of the body
Advanced systems such as liposomes, polymeric nanoparticles and carriers that respond to environmental changes are being actively studied.
13.2.4 Gene Therapy and Genome Editing
Gene therapy is a method for directly targeting resistance mechanisms at the genetic level.
• Silencing genes that cause resistance, such as MDR1
• Restoring function of tumor suppressor genes, such as p53
• Using CRISPR-Cas9 for changes in the genome
These strategies can reverse resistance and increase the effectiveness of cancer treatments.
13.2.5 Immunotherapy and Advanced Cellular Therapies
Immunotherapy is becoming an effective tool for overcoming drug resistance.
• Activating the system to attack resistant cancer cells
• Developing advanced treatments such as CAR-T cells
• Combining immunotherapy with chemotherapy
• These methods help the body recognize and destroy cancer cells those that have developed resistance.
13.2.6 Artificial Intelligence and Machine Learning in Oncology
Artificial intelligence is changing the way cancer research and treatment are done.
• Using datasets to predict resistance patterns
• Optimizing treatment plans
• Discovering drug targets
• Speeding up drug development and clinical trials
AI models help in making decisions based on data.
They improve the accuracy of medicine.
13.2.7 Targeting Tumor Microenvironment (TME)
The tumor microenvironment plays a role in drug resistance.
• Focusing on areas with oxygen and abnormal blood supply
• Changing the behavior of supporting and immune cells
• Helping drugs reach deeper into the tumor tissue
Future treatments aim to disrupt the environment of the tumor.
This makes cancer cells more responsive to treatment.
13.2.8 Biomarker Discovery and Early Detection
Finding biomarkers is important for early detection of resistance.
• Using genomic and proteomic markers
• Liquid biopsies for real-time monitoring
• Predicting treatment response early
This allows for changes in treatment and prevents treatment failure.
13.2.9 Epigenetic Therapy
Changes in the way genes are regulated contribute, to drug resistance.
• Using drugs that block DNA methylation
• Inhibitors of histone deacetylase (HDAC)
• Reversing gene silencing
therapy can restore sensitivity to cancer drugs.
It improves treatment outcomes.
13.2.10 Development of Novel Therapeutic Modalities
therapies are being developed to overcome resistance:
• RNA-based treatments (siRNA, mRNA)
• Proteolysis-targeting chimeras (PROTACs)
• Bispecific antibodies
These innovative methods offer ways to target resistant cancer cells.
REFERENCES

Manish Chaudhari, Sujta Wagh, Mayuri Sonawane, Jayaprada Ghate Drug Resistance in Cancer Therapy: Molecular Mechanisms, Clinical Challenges and Emerging Therapeutic Strategies, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 7, 744-786, https://doi.org/10.5281/zenodo.21160521
 10.5281/zenodo.21160521
10.5281/zenodo.21160521