
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutics, P. Wadhwani College of Pharmacy, Yavatmal, Maharashtra, India.Brix Biopharma Pvt. Ltd., Karad, Maharashtra, India
The present investigation focused on the formulation and evaluation of gastroretentive floating tablets of Valsartan for site-specific drug release. Gastroretentive drug delivery systems (GRDDS) are designed to prolong gastric residence time and improve bioavailability. Floating tablets were prepared using HPMC K100M, Acrypol 971 and Eudragit polymers by wet granulation method. The formulations were evaluated for pre-compression and post-compression parameters including angle of repose, bulk density, hardness, friability and in-vitro drug release studies. FTIR studies confirmed compatibility between drug and excipients. The optimized formulation demonstrated satisfactory buoyancy and sustained drug release characteristics.
Oral drug delivery remains the most widely accepted and preferred route for systemic drug administration owing to its convenience, patient compliance, non-invasive nature, ease of manufacturing, and cost-effectiveness. Despite these advantages, conventional oral dosage forms often exhibit several limitations, including rapid gastric emptying, unpredictable gastrointestinal transit, short biological half-life of drugs, and incomplete drug absorption. These factors may result in fluctuating plasma drug concentrations, reduced bioavailability, and the need for frequent dosing, ultimately affecting therapeutic efficacy and patient adherence.⁷⁻⁹
To overcome these limitations, considerable attention has been directed toward the development of novel oral controlled-release systems capable of prolonging drug residence within the gastrointestinal tract. Among these, gastroretentive drug delivery systems (GRDDS) have emerged as a promising approach for improving the oral bioavailability of drugs that exhibit narrow absorption windows in the upper gastrointestinal tract, are preferentially absorbed in the stomach or proximal small intestine, possess low solubility at intestinal pH, or undergo degradation in the alkaline environment of the intestine. By increasing gastric residence time, GRDDS can enhance drug absorption, reduce dosing frequency, and provide more consistent therapeutic outcomes.¹⁻³˒¹⁰
Several gastroretentive approaches have been investigated, including floating systems, bioadhesive systems, expandable systems, high-density systems, swelling systems, magnetic systems, and raft-forming systems. Among these approaches, floating drug delivery systems (FDDS) are the most extensively studied and commercially successful. FDDS are designed to remain buoyant in gastric fluids due to their lower density compared with gastric contents. Once administered, the dosage form floats on the surface of gastric fluid without significantly affecting the physiological gastric emptying process. As a result, the drug remains in the stomach for a prolonged period, allowing sustained release and improved absorption.⁴˒⁵˒¹¹
Floating drug delivery systems can be broadly classified into effervescent and non-effervescent systems. Effervescent systems utilize gas-generating agents such as sodium bicarbonate and citric acid, which react in acidic gastric media to produce carbon dioxide. The generated gas becomes entrapped within the hydrated polymer matrix, reducing tablet density and enabling buoyancy. Non-effervescent systems rely on swelling and gel-forming polymers that expand upon contact with gastric fluid, thereby maintaining a density lower than that of the surrounding medium. Additional approaches include raft-forming systems, volatile-liquid containing systems, and hollow microspheres.⁵˒¹¹˒¹²
The performance of floating drug delivery systems is strongly influenced by the nature and concentration of matrix-forming polymers. Hydrophilic polymers such as Hydroxypropyl Methylcellulose (HPMC) are widely employed because of their excellent swelling characteristics and ability to form a robust gel barrier that regulates drug diffusion. Carbomer-based polymers such as Acrypol 971 contribute to matrix integrity, swelling capacity, and prolonged drug release. Polymethacrylate derivatives such as Eudragit are also extensively used in controlled-release formulations owing to their favorable physicochemical properties and ability to modulate drug release profiles. The synergistic combination of these polymers enables the development of floating tablets with optimal buoyancy, matrix strength, and sustained-release behavior.⁷⁻⁹˒¹²
Valsartan is a potent and selective angiotensin II receptor blocker (ARB) widely prescribed for the management of hypertension, congestive heart failure, and post-myocardial infarction conditions. The drug exerts its pharmacological action by selectively blocking the binding of angiotensin II to AT₁ receptors, thereby reducing vasoconstriction, aldosterone secretion, and blood pressure. Despite its clinical effectiveness, Valsartan exhibits relatively low aqueous solubility and an absolute oral bioavailability of approximately 23–25%. Furthermore, it possesses a biological half-life of about 6 hours, necessitating repeated administration to maintain therapeutic plasma concentrations. These characteristics make Valsartan a suitable candidate for the development of a gastroretentive sustained-release dosage form.⁶
A gastroretentive floating formulation of Valsartan may offer several advantages, including prolonged gastric residence, controlled drug release, improved dissolution in acidic gastric conditions, enhanced absorption, reduced dosing frequency, and improved patient compliance. By maintaining the dosage form within the stomach for an extended duration, the formulation can provide a continuous release of drug at the absorption site, thereby minimizing fluctuations in plasma concentration and enhancing therapeutic efficacy.⁶˒¹³˒¹⁴
In the present investigation, gastroretentive floating tablets of Valsartan were formulated using HPMC K100M, Acrypol 971, and Eudragit as matrix-forming polymers, along with sodium bicarbonate and citric acid as effervescent agents. The prepared formulations were evaluated for physicochemical characteristics, buoyancy behavior, drug content, in-vitro dissolution profile, and release kinetics. The study aimed to develop an effective floating matrix tablet capable of providing prolonged gastric retention and sustained drug release for improved therapeutic performance of Valsartan.⁶
2. MATERIALS AND METHODS
2.1 Materials
Valsartan (API), HPMC K100M (Yarrow Chem), Acrypol 971 (Corel Pharma Chem), Eudragit (Prayogina Laboratories), lactose, microcrystalline cellulose (MCC), sodium bicarbonate, citric acid, talc, magnesium stearate, croscarmellose sodium. All reagents were of pharmacopeial grade.
2.2 Instrumentation
Analytical balance (RADWAG AS220.R), mechanical sifter (#30, #60), blender (Roto Cube), tablet press (single-punch), UV–Vis spectrophotometer (Thermo Evolution 201), LOD, FTIR (Bruker ALPHA II), tapped density tester (Lab-India TD1025), dissolution tester (Lab-India DS8000+), vernier caliper, hardness tester (Lab-India TH1050S), friabilator (Lab-India FT1020), disintegration tester.
2.3 Method of Preparation of Valsartan Floating Tablets
Wet Granulation Method
Valsartan floating tablets were prepared by the wet granulation technique. Accurately weighed quantities of Valsartan and polymers were mixed thoroughly to obtain a uniform blend. The powder mixture was granulated using a suitable granulating agent until a coherent wet mass was formed. The wet mass was then passed through a sieve to produce granules of uniform size. The resulting granules were dried and resieved to obtain dry granules with desired particle size distribution. The dried granules were blended with lubricants and glidants to improve flow properties and compressibility. Finally, the prepared blend was compressed into tablets using a single-punch tablet compression machine. This method ensured uniform drug distribution, good flow characteristics, and satisfactory tablet quality.
2.3 Preformulation
Description
Valsartan was evaluated for:
Observations
Identification by UV Spectroscopy
The λ max of Valsartan was determined in 0.1 N HC l buffer pH 1.2.
Calibration Curve
Calibration standards were prepared at concentrations ranging from 2–10 μg/mL.
FTIR Study
Drug-excipient compatibility was assessed by Fourier Transform Infrared Spectroscopy (FTIR).
Table 1 : Physical characteristics of Valsartan
|
Parameter |
Observation |
|
Colour |
White |
|
Odour |
Odourless |
|
Taste |
Bitter |
|
Melting point |
116–120 °C |
Table 2 : Solubility studies Valsartan
|
Sr. No |
Solvent |
Solubilty of Valsartan |
|
1) |
Water |
Practically insoluble |
|
2) |
Ethanol |
Freely soluble |
|
3) |
Methanol |
Freely soluble |
The absorption maxima of Valsartan were determined by scanning the sample drug solution concentration in double beam UV spectrophotometer for range of 250nm and standard specification given in Indian pharmacopoeia or literature.
|
Concetration(ug/ml) |
Absorbance(nm) |
|
2 |
0.123 |
|
4 |
0.189 |
|
6 |
0.288 |
|
8 |
0.366 |
|
10 |
0.434 |
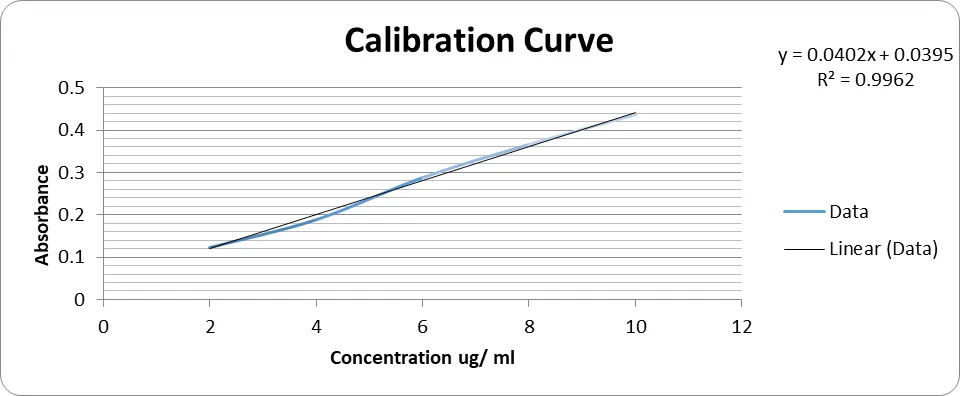
Fig 1 : Standard Calibration curve of Valsartan
Correlation coefficient (R2) = 0.9962
Equation of regressed line = Y = 0.0402x + 0.0395
Where, X = Value of concentration.
Y = Regressed value of Absorbance.
Drug Excipient Interaction Study was carried out to check the interaction between the drug and excipient by using FT-IR spectrophotometer (BRUKER ALPHA - II). Accurately weigh 50 mg of The mixtures of Valsartan formulation and plane drug , were placed separately on the sampling plate of FT-IR spectrophotometer. Then scanning of the sample was performed and IR spectra were obtained as shown in Fig respectively
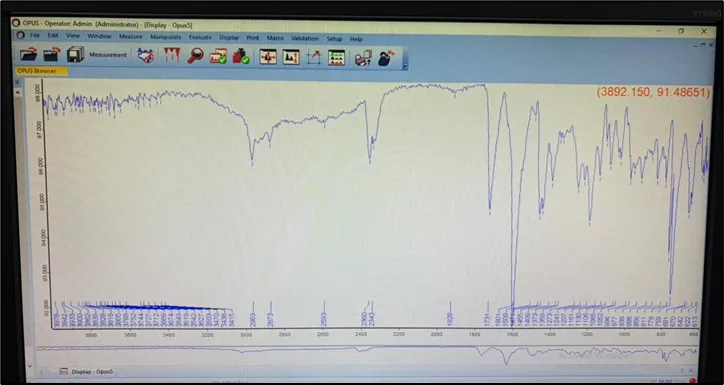
Fig 2: FTIR Interpretation of pure API ( Valsartan )
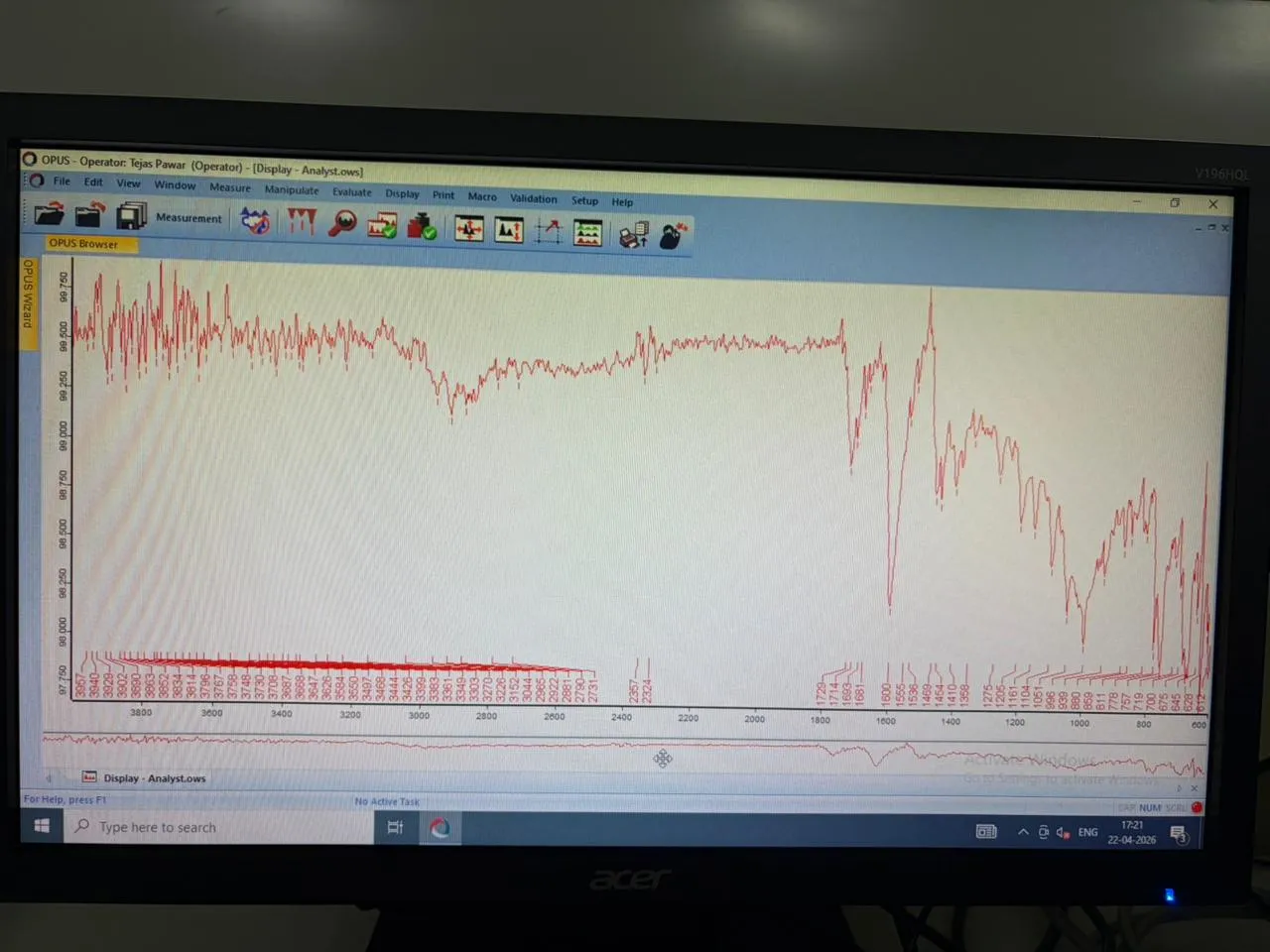
Fig 3: FTIR of API Valsartan and All Excipients
2.4 Tablet Formulation:
Nine formulations (F1–F9; 400 mg/tablet) were prepared by wet granulation. Polymers (HPMC K100M, Acrypol 971, Eudragit) varied across formulations; sodium bicarbonate and citric acid served as effervescent agents. MCC and lactose were diluents; magnesium stearate and talc were lubricant/glidant; croscarmellose was used per design. Wet massing with suitable granulating fluid, screening, drying, resizing, and lubrication preceded compression.
Table 3: Composition of Formulations F1–F9
|
Sr. No. |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
|
Valsartan |
80 |
80 |
80 |
80 |
80 |
80 |
80 |
80 |
80 |
|
Eudragit |
40 |
80 |
40 |
- |
- |
60 |
- |
- |
- |
|
Acrypol 971 |
- |
- |
- |
80 |
- |
- |
60 |
30 |
- |
|
HPMC k 100 M |
40 |
- |
- |
- |
40` |
- |
- |
30 |
80 |
|
Sodium Bicarbonate |
40 |
40 |
40 |
40 |
40 |
40 |
40 |
40 |
40 |
|
Citric Acid |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
|
Microcrytalline Cellulose |
126 |
126 |
166 |
126 |
166 |
146 |
126 |
146 |
126 |
|
Lactose |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
50 |
|
Magnesium Stearate |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
Talc |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
cross Carmellose |
- |
- |
- |
- |
- |
- |
7 |
- |
- |
|
Total |
400 |
400 |
400 |
400 |
400 |
400 |
400 |
400 |
400 |
3. RESULT AND DISCUSSION
3.1 Precompression Evaluation
Angle of Repose (θ) = tan⁻¹ (h/r)
Where:
h = Height of powder heap
r = Radius of powder heap
Bulk Density = Weight of Granules / Bulk Volume
Tapped Density = Weight of Granules / Tapped Volume
Carr's Index (%) = [(Tapped Density − Bulk Density) / Tapped Density] × 100
Hausner's Ratio = Tapped Density / Bulk Density
Table 4 : Micromeritic Properties of Powder Blends Used for Floating Tablet Preparation
|
Batch |
Angle of Repose (°) |
Bulk Density (g/cm³) |
Tapped Density (g/cm³) |
Carr’s Index (%) |
Hausner’s Ratio |
|
F1 |
29.5 ± 0.42 |
0.48 ± 0.01 |
0.58 ± 0.02 |
17.2 ± 0.31 |
1.20 ± 0.02 |
|
F2 |
31.2 ± 0.36 |
0.46 ± 0.02 |
0.57 ± 0.01 |
19.3 ± 0.28 |
1.23 ± 0.01 |
|
F3* |
28.8 ± 0.24 |
0.50 ± 0.01 |
0.60 ± 0.01 |
16.6 ± 0.22 |
1.20 ± 0.01 |
|
F4 |
30.5 ± 0.39 |
0.47 ± 0.01 |
0.59 ± 0.02 |
20.3 ± 0.34 |
1.25 ± 0.02 |
|
F5 |
27.9 ± 0.28 |
0.52 ± 0.02 |
0.61 ± 0.01 |
14.7 ± 0.19 |
1.17 ± 0.01 |
|
F6 |
29.0 ± 0.33 |
0.49 ± 0.01 |
0.60 ± 0.02 |
18.3 ± 0.26 |
1.22 ± 0.02 |
|
F7 |
26.8 ± 0.21 |
0.53 ± 0.01 |
0.62 ± 0.01 |
14.5 ± 0.17 |
1.16 ± 0.01 |
|
F8* |
27.5 ± 0.25 |
0.51 ± 0.01 |
0.61 ± 0.01 |
16.3 ± 0.20 |
1.19 ± 0.01 |
|
F9 |
30.0 ± 0.37 |
0.48 ± 0.02 |
0.59 ± 0.01 |
18.6 ± 0.29 |
1.22 ± 0.01 |
All formulations exhibited satisfactory flow properties, with angle of repose values below 32°, Carr’s index below 21%, and Hausner’s ratio below 1.25. Formulations F3 and F8 showed good flowability and compressibility, indicating their suitability for tablet compression.
3.2 Post compression Evaluation :
The tablets were visually examined for color, surface texture, cracks, capping, chipping, and other physical defects.
Table 5 : Observation of Physical characteristics of the Preformulation
|
Formulation Code |
Colour |
Texture |
Shape |
|
F1 |
White |
Smooth |
Round |
|
F2 |
White |
Smooth |
Round |
|
F3 |
White |
Smooth |
Round |
|
F4 |
White |
Smooth |
Round |
|
F5 |
White |
Smooth |
Round |
|
F6 |
White |
Smooth |
Round |
|
F7 |
White |
Smooth |
Round |
|
F8 |
White |
Smooth |
Round |
|
F9 |
White |
Smooth |
Round |
Table 6 : Post-compression study.
|
Formulation Code |
Hardness (kg/cm²) |
Diameter (mm) |
Thickness (mm) |
Friability (%) |
Weight Variation (mg) |
% Drug Content |
|
|
F1 |
4.75 ± 0.12 |
9.51 ± 0.01 |
4.81 ± 0.03 |
0.47 ± 0.02 |
399.5 ± 1.8 |
|
|
|
F2 |
5.65 ± 0.15 |
9.50 ± 0.01 |
5.22 ± 0.04 |
0.32 ± 0.01 |
399.0 ± 2.1 |
|
|
|
F3* |
5.05 ± 0.08 |
9.51 ± 0.01 |
4.85 ± 0.02 |
0.49 ± 0.02 |
396.5 ± 1.2 |
|
|
|
F4 |
4.85 ± 0.11 |
9.50 ± 0.01 |
5.03 ± 0.03 |
0.32 ± 0.02 |
399.0 ± 2.4 |
|
|
|
F5 |
5.95 ± 0.18 |
9.50 ± 0.02 |
5.47 ± 0.05 |
0.71 ± 0.03 |
399.5 ± 1.4 |
|
|
|
F6 |
6.25 ± 0.21 |
9.51 ± 0.02 |
5.18 ± 0.04 |
0.74 ± 0.04 |
400.0 ± 2.0 |
|
|
|
F7 |
6.15 ± 0.19 |
9.50 ± 0.01 |
5.17 ± 0.03 |
0.56 ± 0.02 |
398.0 ± 1.7 |
|
|
|
F8* |
5.35 ± 0.09 |
9.50 ± 0.01 |
5.01 ± 0.02 |
0.61 ± 0.02 |
399.0 ± 1.3 |
|
|
|
F9 |
6.50 ± 0.22 |
9.52 ± 0.02 |
5.20 ± 0.04 |
0.58 ± 0.03 |
401.0 ± 2.2 |
|
The floating behavior of all formulations (F1–F9) was evaluated by measuring Floating Lag Time (FLT) and Total Floating Time (TFT). The FLT of the formulations ranged from 30 to 50 seconds, indicating rapid buoyancy in the dissolution medium. Formulation F3 exhibited the shortest floating lag time of 30 seconds, whereas F4 showed the highest lag time of 50 seconds. All formulations remained buoyant for more than 12 hours, demonstrating excellent floating ability and prolonged gastric retention. These results confirm that the incorporation of sodium bicarbonate and citric acid effectively generated sufficient carbon dioxide to maintain tablet buoyancy throughout the study period.
Table 7: Floating lag time of prepared tablets
|
Batch |
FLT (sec) |
Total Floating Time (Hrs) |
|
F1 |
45 |
>12 |
|
F2 |
48 |
>12 |
|
F3 |
30 |
>12 |
|
F4 |
50 |
>12 |
|
F5 |
40 |
>12 |
|
F6 |
44 |
>12 |
|
F7 |
38 |
>12 |
|
F8 |
48 |
>12 |
|
F9 |
35 |
>12 |

Fig 4: In vitro buoyancy study showing floating behavior of the optimized gastroretentive floating tablet formulation in 0.1 N HCl
In-vitro drug release studies were carried out using a USP dissolution apparatus containing 0.1 N HCl as the dissolution medium. Samples were withdrawn at predetermined time intervals and analyzed spectrophotometrically. The cumulative percentage drug release was calculated to evaluate the release profile and sustained-release behavior of the formulated gastroretentive floating table
Table 8: In-vitro % drug release profile of batch F1-F9
|
Time (hrs) |
F1 |
F2 |
F3* |
F4 |
F5 |
F6 |
F7 |
F8* |
F9 |
|
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
1 |
10.21 ± 0.42 |
8.34 ± 0.31 |
14.12 ± 0.18 |
8.20 ± 0.29 |
11.08 ± 0.37 |
9.28 ± 0.33 |
8.18 ± 0.26 |
12.40 ± 0.16 |
7.12 ± 0.24 |
|
2 |
18.46 ± 0.58 |
15.27 ± 0.46 |
24.38 ± 0.24 |
14.50 ± 0.41 |
19.22 ± 0.52 |
16.11 ± 0.47 |
15.36 ± 0.39 |
20.60 ± 0.22 |
13.54 ± 0.35 |
|
3 |
26.73 ± 0.74 |
22.41 ± 0.61 |
34.55 ± 0.31 |
19.80 ± 0.55 |
27.64 ± 0.68 |
23.48 ± 0.60 |
22.17 ± 0.53 |
28.90 ± 0.28 |
19.88 ± 0.49 |
|
5 |
40.12 ± 0.96 |
35.36 ± 0.82 |
52.18 ± 0.42 |
32.40 ± 0.75 |
42.27 ± 0.88 |
38.19 ± 0.80 |
36.42 ± 0.71 |
46.20 ± 0.39 |
30.66 ± 0.66 |
|
7 |
55.48 ± 1.12 |
50.27 ± 0.98 |
68.42 ± 0.53 |
46.50 ± 0.91 |
57.36 ± 1.04 |
52.14 ± 0.95 |
50.28 ± 0.86 |
62.60 ± 0.47 |
45.72 ± 0.79 |
|
9 |
68.25 ± 1.28 |
62.18 ± 1.15 |
82.16 ± 0.61 |
58.60 ± 1.03 |
70.18 ± 1.19 |
65.33 ± 1.10 |
63.21 ± 0.97 |
75.30 ± 0.55 |
58.44 ± 0.92 |
|
12 |
80.36 ± 1.42 |
75.14 ± 1.27 |
92.08 ± 0.68 |
75.60 ± 1.18 |
82.25 ± 1.31 |
78.16 ± 1.22 |
76.05 ± 1.10 |
90.30 ± 0.63 |
70.38 ± 1.04 |
All formulations exhibited a gradual increase in cumulative drug release with increasing dissolution time, indicating sustained-release behavior. Drug release after 12 hours ranged from 70.38 ± 1.04% to 92.08 ± 0.68%. Among all formulations, F3 and F8 showed the highest drug release, with 92.08 ± 0.68% and 90.30 ± 0.63% release at 12 hours, respectively, and were considered optimized formulations. In contrast, F4 and F9 exhibited comparatively slower drug release, which may be attributed to higher polymer content and stronger matrix formation. The results demonstrated that the polymer concentration and composition significantly influenced the drug release rate. The low standard deviation values indicated good reproducibility and uniformity of the dissolution studies. Overall, formulations F3 and F8 showed satisfactory sustained-release characteristics and were selected as the optimized batches.
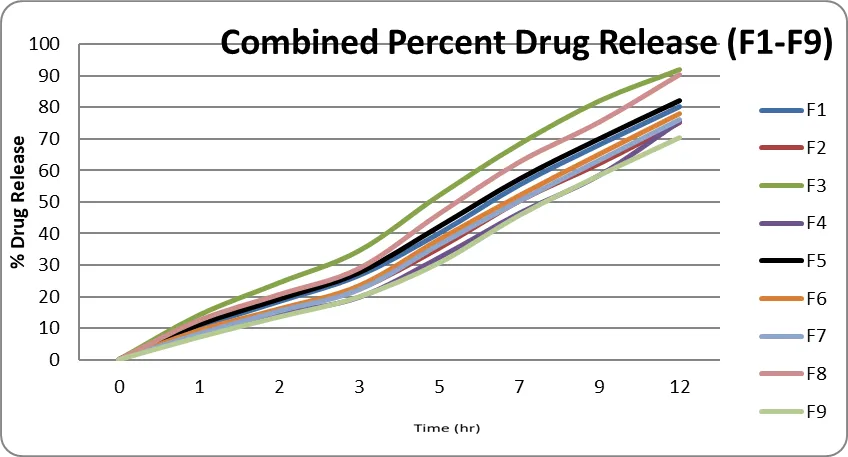
Fig: Cumulative % drug release
Table5: Kinetics of Drug Release
|
Formulation code |
First order plot (R2) |
Zero order plot (R2) |
Higuchi plots (R2) |
Hixon- Crowel cube root plot (R2) |
Korsmeyer-peppas plots |
Best fit model |
|
|
N |
R2 |
||||||
|
F3 |
0.9806 |
0.9716 |
0.9944 |
0.9889 |
0.8145 |
0.9998 |
Korsmeyer-peppas |
|
F8 |
0.9632 |
0.9894 |
0.9718 |
0.9758 |
0.8146 |
0.9967 |
Korsmeyer-peppas |
The release data of formulations F3 and F8 best fitted the Korsmeyer–Peppas model (R² = 0.9998 and 0.9967, respectively). The n values (~0.81) indicate anomalous (non-Fickian) drug release involving both diffusion and polymer relaxation mechanisms.
CONCLUSION
The present study successfully formulated and evaluated a gastro-retentive floating drug delivery system of Valsartan. Preformulation studies confirmed the suitability of the drug for formulation development, while FTIR analysis demonstrated compatibility between the drug and selected excipients. The powder blends exhibited good flow and compressibility characteristics, as evidenced by acceptable micromeritic properties. All tablet formulations complied with pharmacopoeial requirements for post-compression parameters, including hardness, friability, thickness, weight variation, and drug content uniformity. The formulated tablets also showed satisfactory floating behavior and prolonged gastric retention. In vitro drug release studies revealed sustained drug release over 12 hours, with formulations F3 and F8 exhibiting the highest drug release of 92.08% and 90.30%, respectively. Drug release kinetics indicated a controlled release mechanism best described by the Korsmeyer–Peppas model, suggesting anomalous (non-Fickian) diffusion. Overall, the developed gastro-retentive floating tablets of Valsartan demonstrated desirable physicochemical properties, prolonged gastric residence, and controlled drug release, indicating their potential to enhance bioavailability, therapeutic efficacy, and patient compliance.
The authors are thankful to Brix Biopharma Pvt. Ltd. Karad, for providing necessary facilities to carry out the research work.
The authors declare no conflict of interest
REFERENCES

Akshada Fursule, Aasawari Rajgure, Rutika Bawankule,Madhuri Channawar, Mahesh Rao, Formulation And Evaluation of Gastroretentive Drug Delivery System for Site-Specific Drug Release, Int. J. of Pharm. Sci., 2026, Vol 4, Issue 6, 5921-5932, https://doi.org/10.5281/zenodo.20813024
 10.5281/zenodo.20813024
10.5281/zenodo.20813024